


Case Report of Vision Threatening Papilledema due to Idiopathic Intracranial Hypertension
Home / Neuro-Ophthalmology / Grand Rounds Presentations and Cases
Title: Case Report of Vision Threatening Papilledema due to Idiopathic Intracranial Hypertension
Author: Robert Henseler, 4th Year Medical Student, Rutgers University – New Jersey Medical School
CC: Headache and Blurry Vision
HPI: A 20 y/o obese woman presents with a 2-week history of blurry vision and headaches. She was originally diagnosed with a urinary tract infection and prescribed cefuroxime. After seeing her regular physician, she was referred to an ophthalmologist for her visual symptoms where optic nerve swelling on exam and visual field defects on Humphrey Visual Field (HVF) testing were detected. She also complained of neck pain and photophobia so she was sent to the emergency room to rule out the possibility of meningitis. Ophthalmology was consulted and grade 4 disc edema was noted, showing hemorrhages off the discs, tortuous vasculature, and few macular and peripheral hemorrhages. She also had visual field defects on confrontation testing, a left RAPD, and acuity of 20/100 OD and 20/80 OS. A CT and MRI were performed which suggested increased intracranial pressure. A lumbar puncture (LP) was then performed with opening pressure of 56cm H20. At the time of her first LP, 32ml of CSF were removed and closing pressure was 7cm H20. Patient had some resolution of headache and reported slightly improved vision. Her symptoms returned the next day and a repeat LP done the with 30ml of fluid removed.
Testing During Admission:
LP in Lateral Decubitus Position:
- Opening Pressure 56 cmH20
- No signs of infection
MRI Orbits:
- Flattening of the posterior globes
- Enhancement and intraocular protrusion of the optic nerve heads
- Prominent symmetric optic nerve sheath fluid and enhancement
- Mild symmetric proptosis is suggested
MRI Brain:
- No mass lesions
- No hemorrhage
CTV:
- Patent vessels proximally
- Focal narrowing at the bilateral transverse and sigmoid sinus junctions without intraluminal thrombus or evidence of external compression
- Sigmoid sinuses patent distally
The patient was then seen in the neuro-ophthalmology clinic for evaluation.
Neuro-Ophthalmologic History, Exam, and Testing Obtained Day After Admission:
Headache History:
- Located behind her eyes and wrapped around her forehead.
- Started the day following blurry vision and was a sharp pain. 9/10 in severity.
- Originally worse when laying down.
- Pulsatile tinnitus starting 4 days prior to admission.
- Intermittent visual field defects in her left eye when looking straight ahead
- Transient visual obscurations. Her left eye vision was “completely dark” for 2 days prior to admission but it came back on the day before hospital admission.
- Blurry vision in both eyes and constant binocular double vision.
- Tried ibuprofen and acetaminophen but with no relief.
- Morphine given after arrival to the ED, but also provided little to no relief.
- Now after second LP having headaches that are worse when sitting up and better with lying down.
- Patient had no history of migraines, carsickness, nausea, vomiting.
Weight History:
- High School Graduation weight: 170 pounds
- Current weight: Patient thinks 193 pounds but unsure
- Actual weight during admission was 237 pounds
- Maximum weight: Not sure but believes now is her heaviest weight
IIH Associations:
- COPD: Never smoker
- Venous thrombosis: None
- Steroids use and withdrawal: None
- Oral Contraceptive: Nexplanon implant for the past 2 years
- Vitamin A: None
- Tetracyclines: None
- Isotretinoin: None
Exam:
- EOMs were full and ortho bilaterally
- Hertel Testing 20, 20 at base 95
- Visual Acuity OD 20/70 -1, OS 20/70. No improvement with pinhole
- Pupil exam was normal except for a RAPD of 0.6 log in the left eye
- Anterior segment exam was normal
- Fundus exam showed grade 3 disc edema with boggy macula and tortuous vessels OU
Hospital Course:
Patient continued to stay in the Neuro Critical Care Unit. A lumbar drain was placed by neurosurgery after her visit to the ophthalmology clinic. Neuro-ophthalmology continued to follow the patient and it was decided that if she did not have resolution of increased ICP and vision threatening papilledema then a bilateral nerve sheath fenestration would be performed by oculoplastic surgery. Due to lack of improvement the patient underwent surgery on hospital day 3. Her surgery was performed successfully with no complications. Neuro-ophthalmology continued to follow the patient during her stay at the hospital and then manage her care following discharge on hospital day 4. She was discharged on acetazolamide 1000 mg BID to lower CSF production, gabapentin for acetazolamide induced peripheral neuropathy, and hydrocodone/acetaminophen 10/325 for pain.
4 Days after Discharge:
Interval History:
- Headaches improved, occasional headache while upright
- Transient visual obscurations: none
- Pulsatile Tinnitus: resolved
- Vision acuity subjectively improved
- Peripheral Vision: improved
- Diplopia: still had oblique diplopia
Exam:
- Visual Acuity improved to OD 20/40, OS 20/40 -1
- Left RAPD 0.6 log
- Surgical incisions clean with mild upper eyelid edema
- Anterior segment exam normal
- OD grade 3 edema with punctate hemorrhages peripapillary
- OS grade 3 edema (OS>OD) with peripapillary hemorrhage nasally
- Tortuous vessels OU
Plan:
- Continue acetazolamide 1000mg BID
- Follow up 1 week
12 Days after Discharge:
Interval History:
- Patient reports worsening symptoms beginning 5 days prior
- Symptoms include nausea, worsened supine headaches, and more pulsatile tinnitus
- Blurry and double vision subjectively worse
Exam:
- Visual acuity decreased since last visit. 20/60 OD, 20/60+1 OS.
- OD Grade 3 edema, no hemorrhages
- OS Grade 3 edema, superior resolving hemorrhage
Plan:
- Therapeutic LP now
- Increase acetazolamide to 2000mg BID
- Follow up 1 week
- If not improved will consider shunt placement
18 Days after Discharge:
Interval History:
- Visual fields are subjectively the same for her
- Blurry vision the same
- Double vision almost resolved
- Following LP last visit had improvement of symptoms for 1 day
- Pulsatile tinnitus returned
- Headache continues
- Nausea continues
- On acetazolamide 2000mg BID
Exam:
- Visual acuity improved to 20/30-2 OD, 20/30-2 OS. No change with pinhole
- Left RAPD 0.3 log
- OD grade 2 edema, no hemorrhages
- OS grade 2 edema, superior resolving hemorrhage
- Tortuous vessels OU
Plan:
- Still having significant IIH symptoms despite 2000mg BID Diamox
- Side effects almost intolerable
- Visual fields and acuity slightly improved
- Start Lasix 20mg BID trial
- Refer to neurosurgery for shunt placement due to continued IIH symptoms
- Follow up after shunt placement or with worsening symptoms
Discussion of Case:
This was a sudden onset severe case of IIH where many specialties were involved in trying to prevent papilledema induced vision loss. The important aspects of care after admission to the hospital were quick imaging (MRI of the Orbits, Brain and CT Venogram), LP in lateral decubitus, and ophthalmology consultation, examination, and HVF testing. The initially differential diagnosis for increased ICP could include a multitude of pathologic processes.
Differential Diagnosis of Papilledema and Increased ICP:
- Intracranial mass lesion, tumor or hematoma
- Cerebral edema from acute hypoxic ischemic encephalopathy, large cerebral infarction, or traumatic brain injury
- Choroid plexus papilloma causing increased CSF production
- Arachnoid granulation adhesions resulting in decreased CSF absorption
- Obstruction of venous outflow from venous sinus thrombosis or jugular vein compression
- Obstructive hydrocephalus (meningitis, cyst, or Chiari malformation)
- Idiopathic intracranial
The MRI of the orbits, brain, and spine are used to assess for many of these causes as well as the ability to show signs of increased ICP. It is also important to rule out brain herniation which would be a contraindication to LP. The CTV was performed to rule out venous sinus thrombosis. The LP in lateral decubitus position is then used to assess the opening pressure. It is also important to test for infectious causes and meningitis. Quick ophthalmologic consultation is also vital to look at the severity of papilledema and guide management. HVF testing was used to track visual field deficits resulting from papilledema and OCT was used to measure and follow the amount of optic nerve swelling. After all the testing, as well as obtaining a thorough history, the diagnosis of IIH was made with weight gain and obesity as the most likely etiology.
The rapid progression and severity of symptoms in the patient’s clinical course led to more aggressive treatment of her vision threatening papilledema. During her first 2 days in the hospital she had two LPs performed and a drain placed to remove fluid and control pressure. The decision for bilateral optic nerve sheath fenestration—while controversial due to risk of damage to the optic nerve—can be safely performed by a well-trained oculoplastic surgeon and provide significant relief to the optic nerves. Performing optic nerve sheath fenestrations unilaterally is more common. During the case, discussions were made about how best to perform optic nerve sheath fenestration while a patient has a drain. Having fluid surrounding the optic nerve allows for easier and safer surgery but increases the pressure and can cause more damage to the optic nerve. It was decided that moving forward it is best to place the drain at a height so as to maintain a pressure of 20 cmH20 a few hours prior to surgery thus allowing safe surgical approach while minimizing the risk of progressive visual damage.
Since the patient continued to have severe symptoms of increased ICP as well as minimal improvement of OCT and HVF the dose of her acetazolamide was increased to 2000mg BID at day 12 post D/C. This is a large dose and the patient was unable to tolerate the adverse effects of the medication and still continued to have symptoms of increased ICP. While not usually necessary to control optic nerve swelling and IIH symptoms, the decision was made to place a CSF shunt. This was a severe and rapid case of IIH. Aggressive medical and surgical treatment was used in order to minimize optic nerve swelling and symptoms of increased ICP. In summary, severe visual symptoms should always be addressed rapidly by an ophthalmologist as delay can lead to permanent visual deficits. Management should always be individually tailored as not all cases require the same treatment course.
Fundus Photo Right – 4 days post D/C

Fundus Photo Left – 4 days post D/C
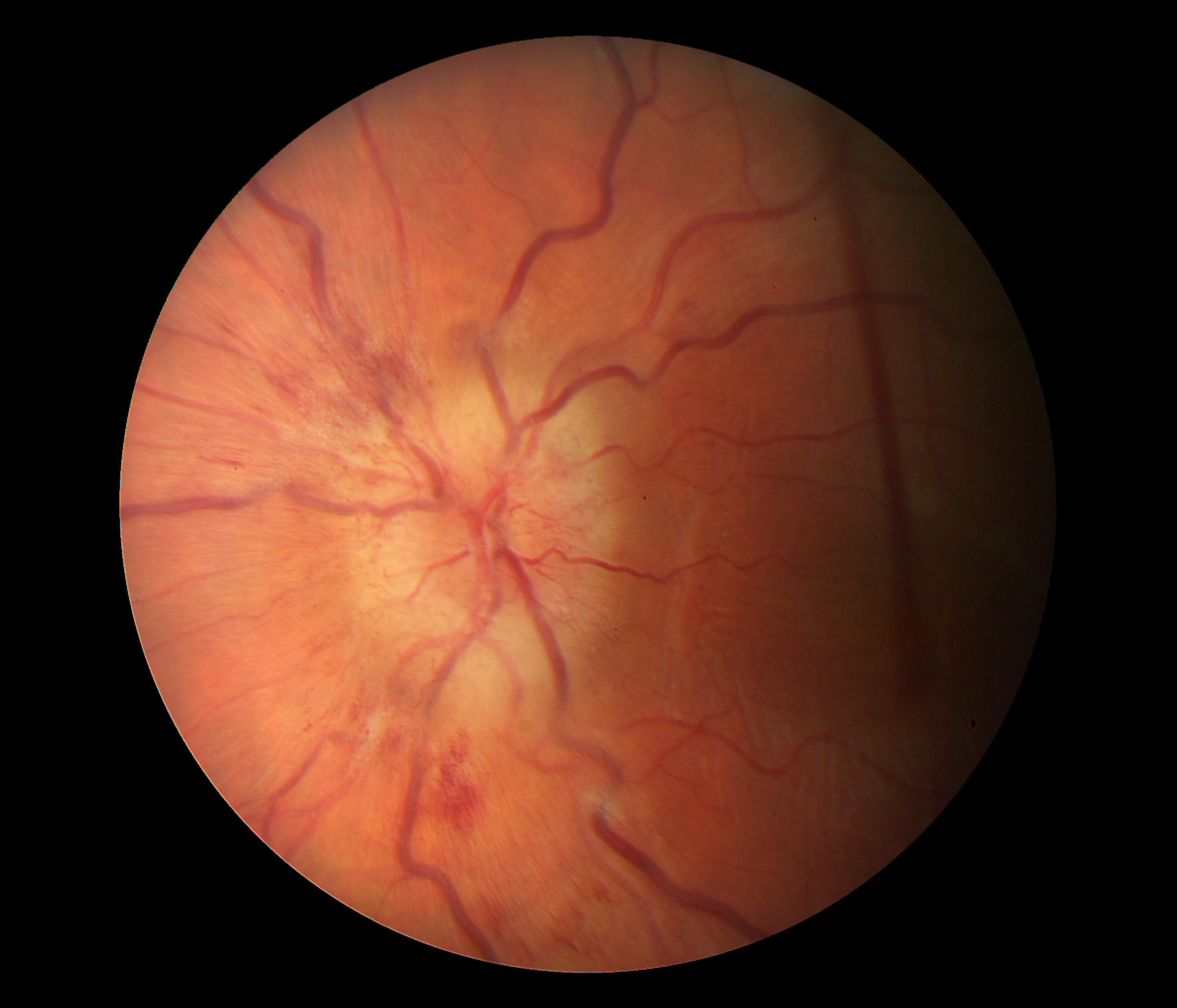
Grade 3 papilledema OU with loss of major vessels bilaterally as they exit the disc. The left disc is more edematous than the right. Vessels are tortuous and areas of hemorrhage are seen.
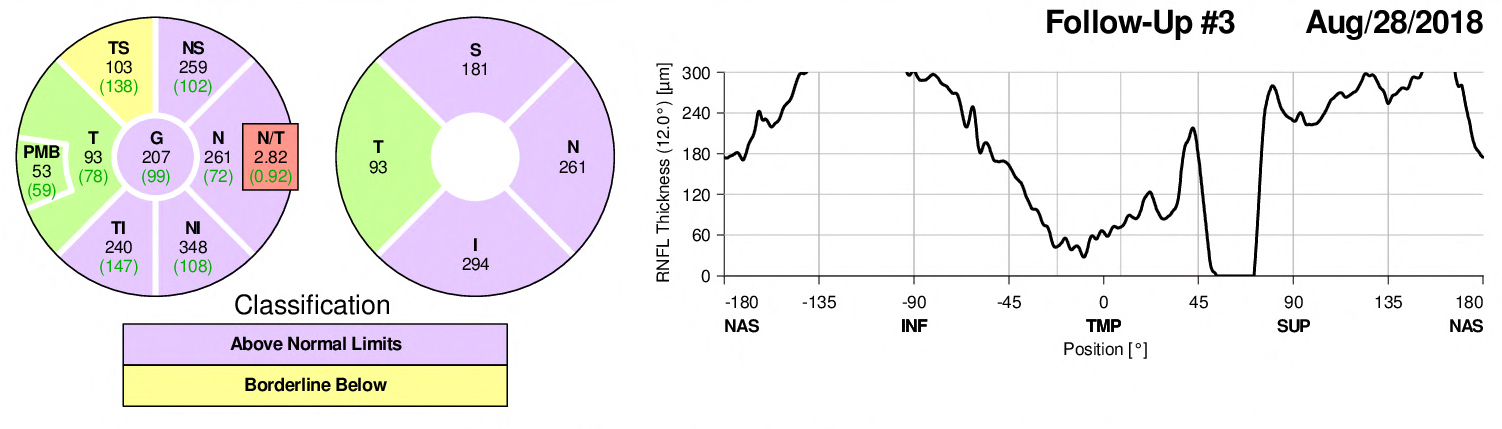
OCT Right – during admission

OCT Right – 4 days post D/C

OCT Right – 12 days post D/C
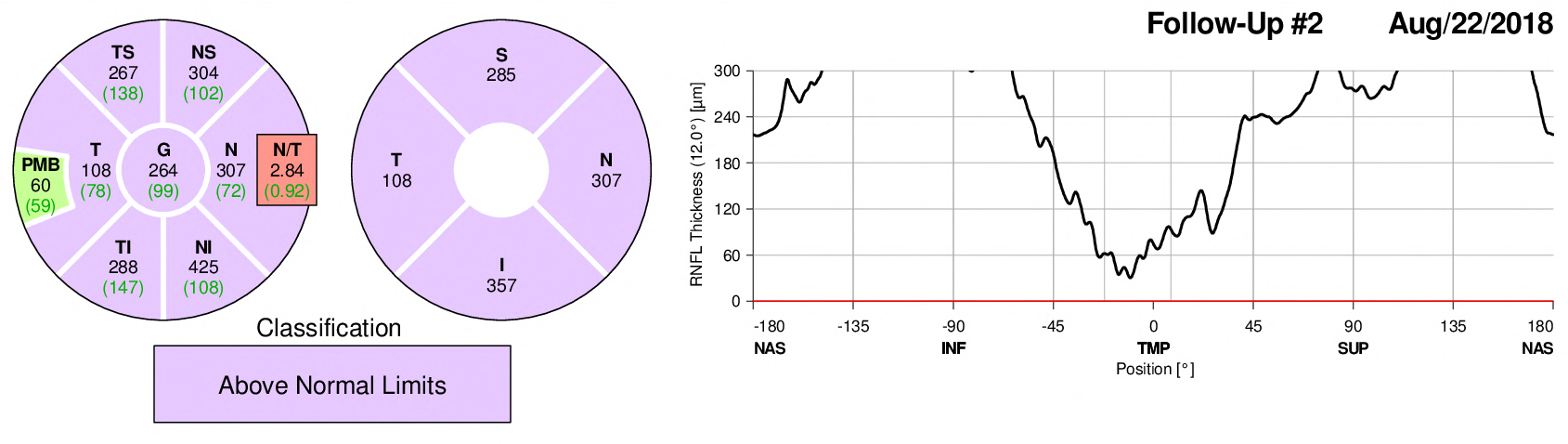
OCT Right – 18 days post D/C
The edema of the right optic nerve progresses to week day 12 and then begins to resolve slightly by day 18 even though measurements are still above normal limits.
OCT Left – during admission

OCT Left – 4 days post D/C

OCT Left – 12 days post D/C

OCT Left – 18 days post D/C

Optic nerve swelling is more significant in the left eye compared to the right which corresponds to the left RAPD as well as exam findings. It is slightly improved from baseline after 2 LPs, a drain, and optic nerve sheath fenestration, but then progresses again on day 12. By day 18 after 1 week of 2000mg BID acetazolamide it is resolving slightly.
HVF Right – during admission

HVF Right – 4 days post D/C
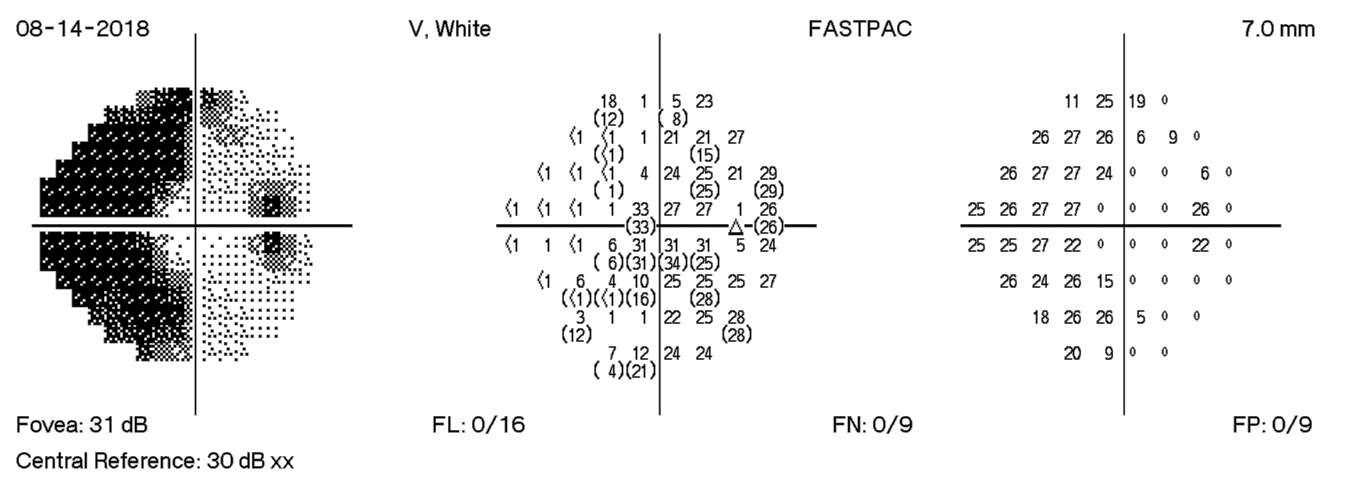
HVF Right – 12 days post D/C

HVF Right – 18 days post D/C

The HVF on the right shows enlarge blind spots temporally and arcuate defects nasally. There is progression of visual field losses until day 12 post D/C with some improvement by day 18 post D/C. These findings correspond with patient symptoms as well as with OCT measurements.
HVF Left – during admission
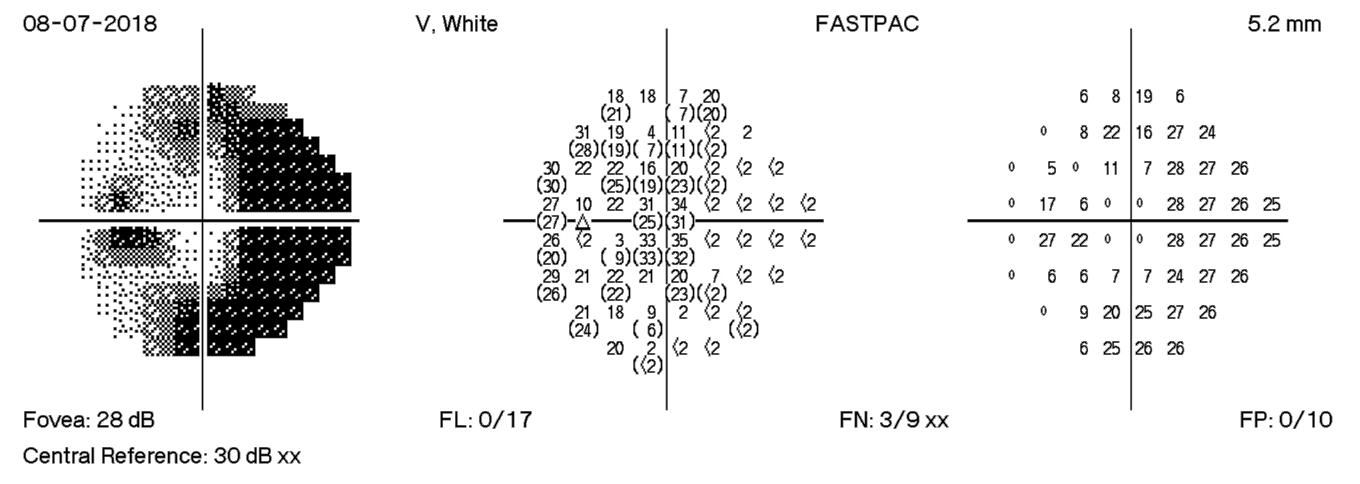
HVF Left – 4 days post D/C

HVF Left – 12 days post D/C
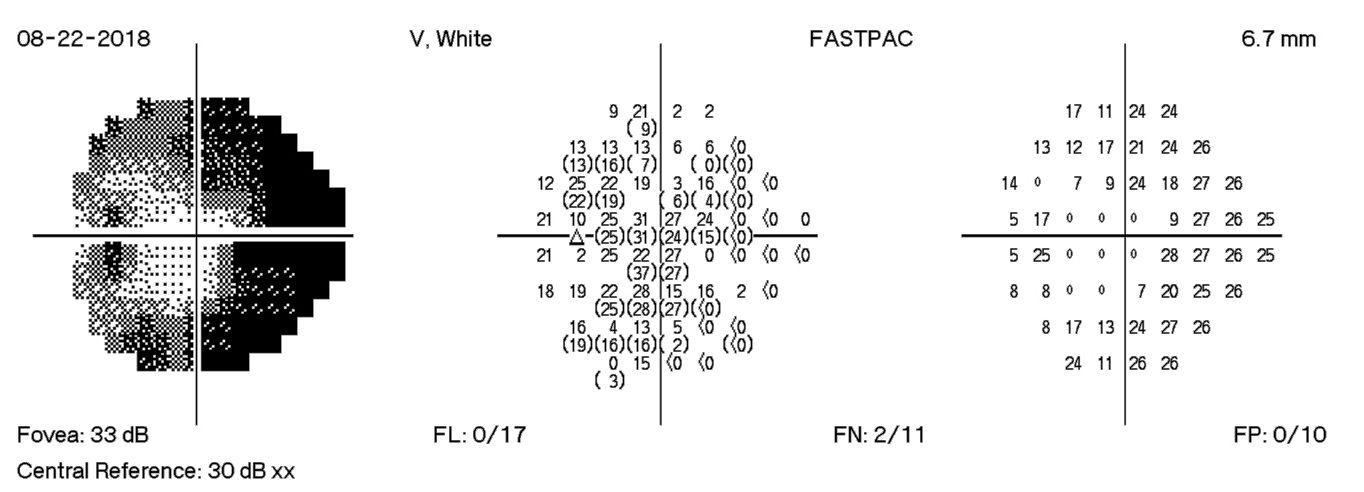
HVF Left – 18 days post D/C

The HVF on the left shows enlarge blind spots temporally and arcuate defects nasally that expand past midline temporally. There is progression of visual field losses until day 12 post D/C with some improvement by day 18 post D/C. The visual field defects are more significant in the left eye than the right eye. These findings correspond with exam findings, fundus photographs, and OCT measurements.
Identifier: Moran_CORE_25503
Case Report of Idiopathic Intracranial Hypertension and Frisen Scale Papilledema Grading
Home / Neuro-Ophthalmology / Grand Rounds Presentations and Cases
Title: Case Report of Idiopathic Intracranial Hypertension and Frisen Scale Papilledema Grading
Author (s): Cole Swiston MSIV, Meagan Seay MD
Photographer: Danielle Princiotta, Becky Weeks
Date: 09/17/18
Image or video:

Figure 1. Color fundus photos obtained during initial examination. A) Grade 4 papilledema in the patient’s right eye, marked by circumferential disc elevation, and obscuration of vessels both on the disc and leaving the disc. Disc hemorrhages are also present. B) Grade 4 papilledema in the left eye with disc hemorrhages.
Keywords/Main Subjects: papilledema, idiopathic intracranial hypertension, IIH, papilledema grading
CORE Category:
http://morancore.utah.edu/section-05-neuro-ophthalmology/
Subcategory: Decreased Vision > Optic Neuropathy
Diagnosis: Idiopathic Intracranial Hypertension
Description of Image:
History of present illness: The patient is a 27-year-old woman, 5 ½ weeks pregnant, who presented to the neuro-ophthalmology clinic with a one-week history of peripheral vision loss in both eyes. She also experienced a left sided throbbing headache, pain behind her eye, and associated neck pain. She had nausea and vomiting during the same time frame. The patient originally presented to an urgent care facility 2 days prior, where the physician noted possible papilledema on exam and transferred her to the emergency room for evaluation. In the ER, the patient underwent lumbar puncture with an opening pressure of 56 cm H2O. CSF glucose, protein, and WBCs were within normal limits. She then underwent an MRI of her head which revealed no structural lesions, acute hemorrhage or infarct though this did show signs of elevated intracranial pressure including bilateral flattening of the posterior sclera and an “empty sella”. An MRV was also performed which showed no evidence of venous outflow obstruction. She was prescribed Diamox but did not begin taking the medication out of fear that it would harm her baby. She had no diplopia, flashers, or floaters in her vision, and had no history of blood clots, though she had 4 prior miscarriages. She was taking an 81 mg aspirin per the recommendation of her obstetric provider and had been on progesterone for the last three weeks during this pregnancy. The patient had no history of doxycycline, tetracycline, steroid, lithium, or Vitamin A derivative use. The last time she was on an oral contraceptive was eight years ago. She weighed 220 pounds and her weight was stable for the past year.
Neuro-ophthalmology examination: The patient’s visual acuity was 20/20 in each eye; her pupils were reactive without a relative afferent pupillary defect. Extraocular movements were full in both eyes, and visual fields performed by confrontation revealed partial inferonasal defect in the right eye. She performed Humphrey 24-2 Visual Field testing which revealed significant peripheral field loss in both eyes, the left eye more so than the right. She identified 11.5/15 Ishihara plates correctly in the right eye, 12.5/13 in the left eye. Slit lamp exam of the anterior segment was unremarkable, and fundus exam revealed grade 4 papilledema in both the right and left (Figure 1) with mild vascular tortuosity. OCT-RNFL confirmed bilateral optic disc swelling.
Clinical Course: The patient was diagnosed with papilledema secondary to idiopathic intracranial hypertension (IIH). After discussing with maternal fetal medicine, it was decided that the benefits of Diamox (Pregnancy Category C) outweighed the risk and the patient agreed to begin taking 500 mg twice a day, increasing to 1000 mg twice a day after two days. Given the severity of her papilledema and visual field defects, the patient was admitted for lumbar drain placement, at which time her opening pressure was 32 cm H2O. The lumbar drain was removed after four days in the hospital. On the day of drain removal, the patient was discharged and seen for follow-up in neuro-ophthalmology clinic. At that time, her visual acuity remained 20/20 in each eye. Papilledema was still present on fundus exam and by RNFL but improved in both eyes, and her objective visual field testing (Humphrey 24-2) showed significantly improved peripheral defects. The patient was continued on Diamox 1000 mg twice a day and was scheduled for close follow up for management of this new diagnosis of IIH.
Discussion: Idiopathic intracranial hypertension (IIH), also known as pseudotumor cerebri, is a condition of increased intracranial pressure (ICP) of unknown etiology. The disease primarily affects obese women of child bearing age, but other risk factors include obstructive sleep apnea, hypothyroidism, anemia, autoimmune conditions, and specific medication use. These medications include steroids, lithium, oral contraceptives, tetracyclines, and vitamin A derivatives. Common non-ocular symptoms include headache, nausea, vomiting, and pulsatile tinnitus (the sensation of blood flow and “whooshing” in the ears). Ocular symptoms are variable, but generally include visual disturbance, either transient episodes of visual loss with position changes or Valsalva maneuver, or peripheral field defects. Central vision is usually spared until very late in the disease. Diplopia may occur if increased intracranial pressure leads to a unilateral or bilateral sixth cranial nerve palsy.1 The main ocular sign of IIH and increased ICP is papilledema, characterized by bilateral optic disc swelling, elevation, blurring of the disc margins, and obscuration of optic disc blood vessels. The modified Frisén scale has been used to grade the severity of papilledema, summarized in Figure 2.2 If IIH is suspected based on presenting clinical symptoms, the first step in diagnosis is fundoscopy to assess for papilledema. Optical coherence tomography (OCT) can be used to quantify the optic disc swelling, and visual field testing is useful to assess for the degree of peripheral field loss. The next steps in diagnosis involve establishing objective evidence of increased ICP and ruling out other potential etiologies. This is accomplished with neuroimaging, usually an MRI, which excludes structural causes (i.e. mass lesions, hemorrhage, or obstructive hydrocephalus), and MRV to rule out venous outflow obstruction (i.e. dural sinus thrombosis). A lumbar puncture with opening pressures confirms elevated ICP and evaluates for alternative etiologies of elevated ICP, including infection, inflammation, or neoplasm.3 Based on these results, the modified Dandy criteria can be used to establish a diagnosis of IIH:
- Signs and symptoms of increased ICP
- Absence of localizing findings on the neurologic exam, other than sixth nerve palsy
- Awake and alert patient
- Normal neuroimaging findings except for signs of elevated ICP
- Increased CSF opening pressure (>25 cm H2O) with normal CSF composition
- No other cause of elevated ICP found4
The management of IIH consists of both medical and surgical/intervention options depending on the severity of papilledema and symptoms. The mainstay of treatment consists of weight reduction and medical management, usually with acetazolamide. Other diuretics including furosemide are sometimes used alone or in combination with acetazolamide. Interventional options range from large volume lumbar punctures and lumbar drains, which are more temporary measures, to lumboperitoneal (LP) or ventriculoperitoneal (VP) shunts, venous sinus stenting, and optic nerve sheath fenestrations.3

Figure 2. Modified Frisén scale for grading of papilledema. Key features of each grade are marked with an asterisk (*).
References:
- Markey KA, Mollan SP, Jensen RH, Sinclair AJ. Understanding idiopathic intracranial hypertension: mechanisms, management, and future directions. Lancet Neurol. 2016;15(1):78-91. doi:10.1016/S1474-4422(15)00298-7
- Diagnosis and Grading of Papilledema in Patients With Raised Intracranial Pressure Using Optical Coherence Tomography vs Clinical Expert Assessment Using a Clinical Staging Scale | Neuro-ophthalmology | JAMA Ophthalmology | JAMA Network. https://jamanetwork.com/journals/jamaophthalmology/fullarticle/425762. Accessed September 15, 2018.
- Jensen RH, Radojicic A, Yri H. The diagnosis and management of idiopathic intracranial hypertension and the associated headache. Ther Adv Neurol Disord. 2016;9(4):317-326. doi:10.1177/1756285616635987
- Friedman DI, Liu GT, Digre KB. Revised diagnostic criteria for the pseudotumor cerebri syndrome in adults and children. Neurology. 2013;81(13):1159-1165. doi:10.1212/WNL.0b013e3182a55f17
Faculty Approval by: Griffin Jardine, MD
Footer: Copyright statement: Copyright Cole Swiston, ©2018. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
Disclosure (Financial or other): The authors have no financial disclosures.
Identifier: Moran_CORE_25475
Custom Implant for Correction of Enophthalmos After Orbital Fracture Repair
Home / Orbit, Eyelids, and Lacrimal System / The Anophthalmic Socket
Title: Custom Implant for Correction of Enophthalmos After Orbital Fracture Repair
Author: Benjamin West, MSIV, Loma Linda University
Date: 7/24/2018
Image or video:

Figure 1. CT scan at admission demonstrating right medial wall blow-out fracture as well as extensive damage to the right globe.

Figure 2. CT scan at 5 months after enucleation and initial fracture repair showing significant right sided enophthalmos and persistence of medial orbital wall fracture.
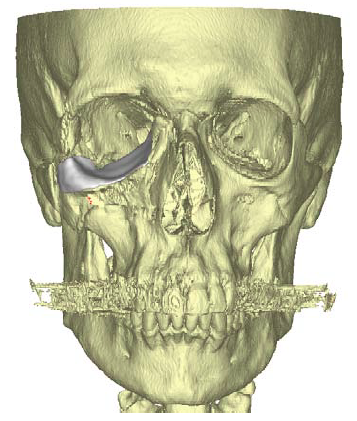
Figure 3. 3D virtual reconstruction of patient anatomy with custom porous polyethylene implant in place.
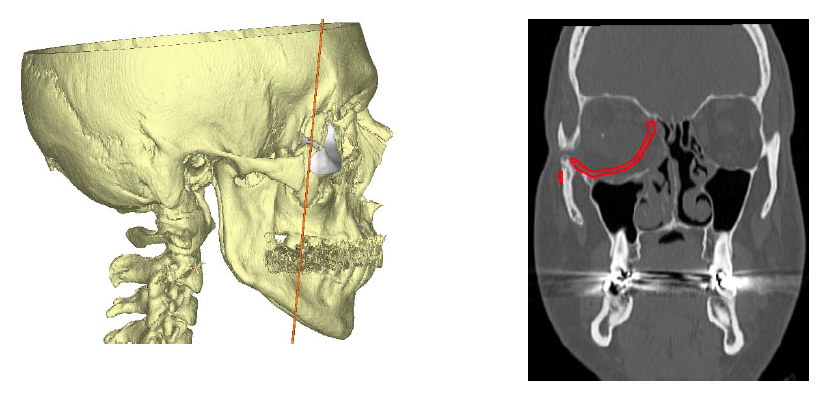
Figure 4. CT scan showing proposed position of custom porous polyethylene implant and subsequent reduction of orbital volume to correct right-sided enophthalmos.
Keywords/Main Subjects: Orbital fractures, Le Fort fractures, Open Globe, Enophthalmos, Orbital Implant; Porous Polyethylene; Custom Implant
CORE Category: Orbit, Eyelids and Lacrimal System > The Anophthalmic Socket > 4. Orbital Implants > “Custom Implant for Correction of Enophthalmos After Orbital Fracture Repair: Case Report”
Diagnosis: Enophthalmos after orbital fracture repair
Description of Image:
This is a 40 year old male who presented to the emergency department after being struck by a heavy chain in the face at work. Initial examination showed extensive facial lacerations (brow, nose, eyelid and temple) as well as a 1 cm laceration of the right cornea and sclera with expulsion of orbital contents. CT scans at admission showed hemi-Le Fort fractures 1, 2 and 3 on the right side, with a zygomaticomaxillary complex fracture and fracture of all four orbital walls (Figure 1). The left side exhibited a hemi-Le Fort 2 fracture, as well as medial and inferior orbital wall fractures.
Due to the extensive damage to the globe, the patient was subsequently taken to the operating room for enucleation and implantation of an 18 mm porous polyethylene implant by oculoplastics. Plastic surgery completed the facial fracture repair. Floating zygoma fractures were plated and anchored to the frontal bone and the right orbital floor was plated with resorbable material to contain the orbital implant in normal position.
At 5 months post-op the patient was noted to have significant right-sided enophthalmos > 2 mm, as well as a severely sunken superior sulcus. Repeat imaging showed osseous bridging of the majority of facial fractures, but persistent right orbit medial blowout fracture with medial herniation of orbital contents and irregularity of the right orbital floor (Figure 2). At this time the patient was agreeable to undertake enophthalmos repair of the right eye with implantation of a customized porous polyethylene implant.
Fine-cut updated CT images were sent to Stryker where a virtual reconstruction plan was made according to the imaging and surgeon specifications. The orbital implant was made from porous polyethylene using a 3D printer and tailored specifically to the anatomy of the patient (Figures 3 and 4).
The patient was taken to the operating room with oculoplastics where an incision was made in the inferior fornix of the right lower eyelid. Dissection was carried out to the inferior orbital rim with subsequent elevation of the periosteum and periorbita. The custom implant was then inserted into the orbit and positioned to correct the enophthalmos as compared to the left eye. The implant was screwed into place at the inferior orbital rim.
At the following post-op examination significant improvement was noted in the enophthalmos and sunken superior sulcus of the right eye with high patient satisfaction. Mild ptosis was noted of the right eye and the patient was counseled on possible future repair if unimproved.
Faculty Approval by: Doug Marx
Footer:
- Copyright Moran Eye Center, ©2018. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Disclosure (Financial or other):
None
Peripheral Leakage, Avascularity, and Non-perfusion –A Case of Familial Exudative Vitreoretinopathy
Home / Retina and Vitreous / Congenital and Developmental Abnormalities
Title: Peripheral Leakage, Avascularity, and Non-perfusion – A Case of Familial Exudative Vitreoretinopathy
Author (s): Blake H. Fortes, MSIV, Florida International University Herbert Wertheim College of Medicine
Photographer: Moran Eye Center
Date: 6/27/2018
Image or video:

Image 1: Montage color fundus photograph of the right eye demonstrating 1) a vitreous adhesion to the optic nerve with temporal macular traction, 2) vascular dragging and tortuosity, 3) far peripheral fibrotic changes overlying atrophic and pigmentary changes, and 4) exudates in the temporal periphery.

Image 2: Montage color fundus photograph of the left eye demonstrates a relatively normal fundus with some slight vascular tortuosity in the temporal periphery.

Image 3: Fluorescein angiogram of the right eye demonstrates multiple peripheral areas of focal hyperfluorescence that were shown to increase in intensity in the late phase along with diffuse leakage in the periphery and temporal peripheral non-perfusion.
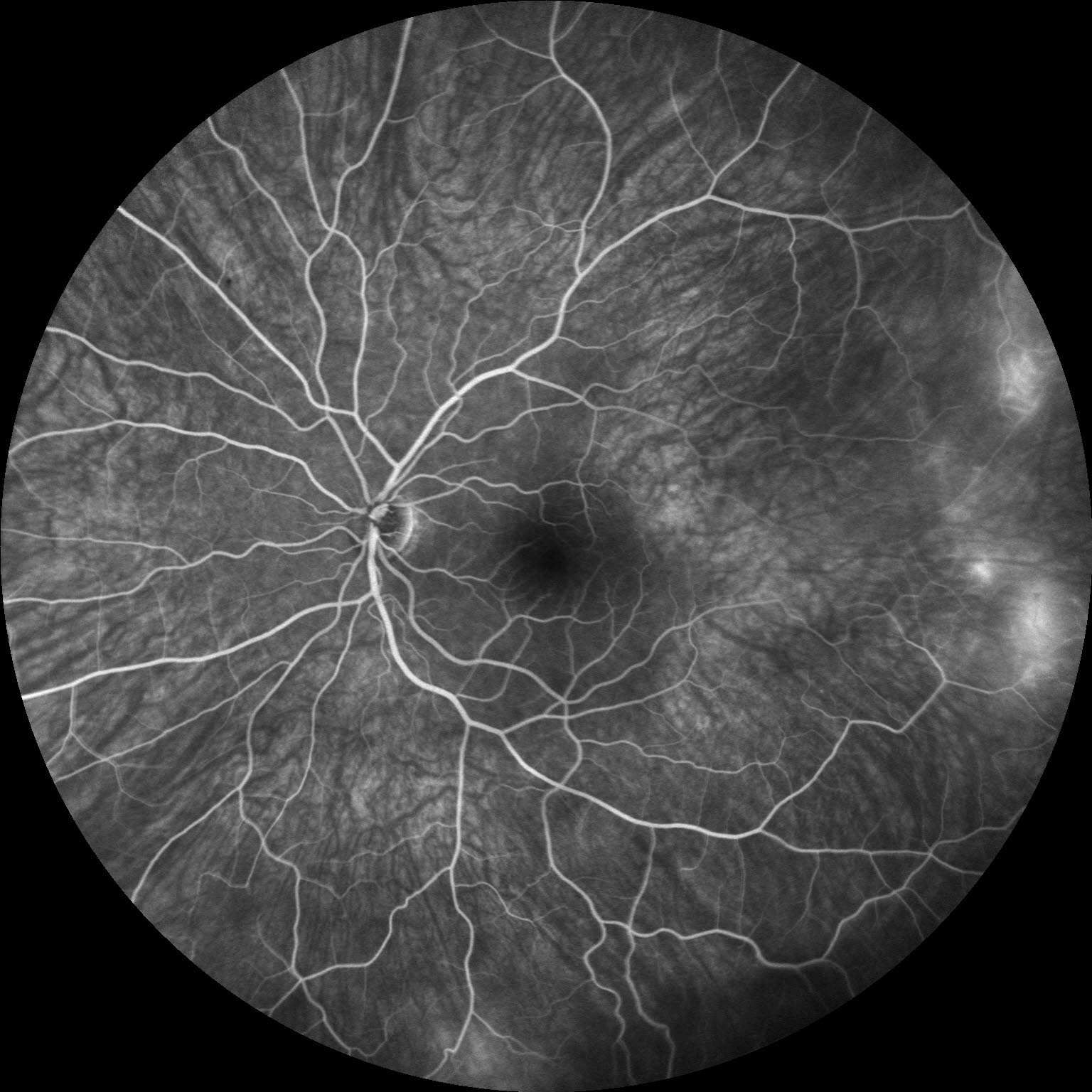
Image 4: Fluorescein angiogram of the left eye revealing multiple areas of temporal vascular leakage with a broad temporal area of non-perfusion, which illustrates, not only, the importance of wide-field fluorescein angiography for diagnosing familial exudative vitreoretinopathy, but also the disease asymmetry that is characteristic of FEVR.
Keywords/Main Subjects: Familial exudative vitreoretinopathy, FEVR, peripheral avascularity, leakage, non-perfusion, neovascularization
Secondary CORE Category: Pediatric Ophthalmology and Strabismus / Disorders of the Retina and Vitreous
Diagnosis: Familial Exudative Vitreoretinopathy
Summary of Case: Patient is a 21 year old female with a diagnosis of a vasoproliferative tumor in the right eye who noted sudden onset of painless drastic decreased visual acuity in the right eye, which had drastically worsened over the last two months and was accompanied by floaters. She denied any photopsias. She has a history of myopia, and has always noticed decreased visual acuity in the right eye. She has no history of eye trauma, or surgery and was born at term, has a normal developmental history, and denied supplemental oxygen use at birth. Family ocular history was significant for a grandmother who had a retinal detachment requiring multiple surgeries. On exam, her visual acuity with correction in the right eye was 20/125 and in the left eye was 20/30 and was noted to have exotropia of the right eye. Her dilated fundus exam in the right eye revealed a tilted, small optic nerve with a vitreal adhesion from the disc to a temporal scar along with macular edema, temporal macular traction, epiretinal membrane, vascular dragging and tortuosity, as well as a fibrotic white lesion at 10 o’clock, surrounded by retinal pigment epithelial changes, and nearby exudates.
Familial Exudative Vitreoretinopathy:
FEVR is a disorder characterized by incomplete vascularization of the peripheral retina typically due to mutations in the Wnt signaling pathway, which is involved in organogenesis and angiogenesis of the eye. These gene mutations include NDP, FZD4, LRP5, and TSPAN12. Novel mutations in ZNF408 and KIF11 have recently been elucidated, but are not involved in the Wnt signaling pathway. The inheritance pattern varies depending on the mutation and may range from autosomal dominant (most commonly), to autosomal recessive, to X-linked recessive or even sporadic, as only 20-40% of patients with FEVR have a positive family history. Therefore, a negative family history does not exclude this diagnosis. This condition is characterized by variable expressivity and asymmetric disease.
The hallmark and most common finding of FEVR is avascularity in the temporal periphery of the retina with associated retinal neovascularization and fibrosis at the junction between the vascular and avascular retina. This fibrosis may result in traction of the macula and retinal vessels, resulting in macular dragging and radial retinal folds. Macular dragging may result in exotropia, as illustrated in this patient. Subretinal exudation, and any type of retinal detachment (rhegmatogenous, tractional, and exudative) may occur as well. Other less common findings associated with this disorder include secondary epiretinal membrane formation, vitreous hemorrhage, secondary glaucoma (neovascular or phacomorphic), retained hyaloid vascular remnants, and persistent fetal vasculature.
Differential diagnosis includes retinopathy of prematurity, Coats’ disease, Norrie’s disease, osteoporosis pseudoglioma syndrome, incontinentia pigmenti, persistent fetal vasculature, vasoproliferative tumor, and ocular toxocariasis.
The staging for FEVR includes:
- Stage 1: avascular periphery
- Stage 2: retinal neovascularization without exudate (A) or with exudate (B)
- Stage 3: extramacular retinal detachment without exudate (A) or with exudate (B)
- Stage 4: subtotal macula-involving retinal detachment without exudate (A) or with exudate (B)
- Stage 5: total retinal detachment
Only patients who have progressed significantly or are at high risk of progression should be treated. In stage 1-2A disease, the avascular retina should be treated with laser photocoagulation to decrease complications related to retinal neovascularization. Retinal detachment should be treated surgically via pars plana vitrectomy, scleral buckle, or a combination of these two approaches. Retinal exudation and neovascularization may also be managed adjunctively via intravitreal anti-VEGF injection prior to surgery. Due to the unpredictable course of FEVR, lifelong monitoring is indicated. Examination of family members is also warranted to reveal previously undiagnosed cases of FEVR.
Format: Case Presentation
References:
- Chen K, Wang N, Wu W. Familial Exudative Vitreoretinopathy. JAMA Ophthalmol. 2017;135(4):e165487. doi:10.1001/jamaophthalmol.2016.5487.
- Gilmour DF. Familial exudative vitreoretinopathy and related retinopathies. Eye. 2015;29(1):1-14. doi:10.1038/eye.2014.70.
- Natung T, Venkatesh P, Thangkhiew L, Syiem J. Asymmetric presentations of familial exudative vitreoretinopathy. Oman Journal of Ophthalmology. 2013;6(2):129-130. doi:10.4103/0974-620X.116661.
- Ranchod TM, Ho LY, Drenser KA, Capone A, and Trese MT. Clinical presentation of familial exudative vitreoretinopathy. Ophthalmology. 2011;118(10):2070-2075.
- Shastry, B. S. (2009), Persistent hyperplastic primary vitreous: congenital malformation of the eye. Clinical & Experimental Ophthalmology, 37: 884-890. doi:10.1111/j.1442-9071.2009.02150.x
- Shields CL, Kaliki S, Al-Dahmash S, et al. Retinal Vasoproliferative TumorsComparative Clinical Features of Primary vs Secondary Tumors in 334 Cases. JAMA Ophthalmol. 2013;131(3):328–334. doi:10.1001/2013.jamaophthalmol.524
- Sızmaz S, Yonekawa Y, T. Trese M. Familial Exudative Vitreoretinopathy. Turkish Journal of Ophthalmology. 2015;45(4):164-168. doi:10.4274/tjo.67699.
- Tauqeer Z, Yonekawa Y. Familial exudative vitreoretinopathy: Pathophysiology, diagnosis, and management. Asia Pac J Ophthalmol (Phila). 2018;7(3):176-182.
Faculty Approval by: Dr. Albert Vitale
Footer:
- Copyright statement: Copyright Author Name, ©2016. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
- Attribution/citation suggestions
Disclosure (Financial or other): None
Hydrogel Intraocular Lens Opacification and Calcification Pathology using a MemoryLens Model
Home / Lens and Cataract / Complications of Cataract Surgery
Title: Hydrogel Intraocular Lens Opacification and Calcification Pathology using a MemoryLens Model
Author(s): Jed H Assam, M.S., and Nick Mamalis, MD
Photographer: Jed H Assam
Date: 7/13/2016
Location in Core: Lens and Cataract > Complications of Cataract Surgery > Complications of IOL Implantation
Keywords / Main Subjects: IOL Calcification, IOL Opacification, MemoryLens, Hydrogels, Hydrophilic Acrylic
Diagnosis / Differential Diagnosis: Posterior Capsular Opacity, Soemmering’s Ring, Anterior Capsule Contraction Syndrome (ACCS)/Capsular Phimosis, Anterior Vitreous Floaters

Figure 1. The anterior surface of an explanted 3-piece hydrophilic acrylic IOL (MemoryLens) with significant calcification shown using light microscopic (large) and stereotactic (small) imaging.
Background
Intraocular lens (IOL) opacification and calcification represent uncommon, but noteworthy causes of blurry vision and decreased visual acuity in pseudophakic patients. Awareness of this pathology becomes particularly important when considering the consequences of reflexively performing initial, errant procedures (nd:YAG and vitrectomy) directed at more common anatomic sources of visual disturbance in pseudophakic populations that typically includes the capsular bag or hyaloid.1,3 A survey (n = 142) evaluating foldable IOL complications requiring removal or secondary interventions identified IOL opacification as a minor cause of postoperative complication in most lens categories evaluated.6 However, for hydrogel (hydrophilic acrylic) IOLs, which represented 4% of the IOLs used in the study, post-operative opacification/calcification was the most common reason for lens removal.6
An explanted, calcified, posterior chamber IOL, MemoryLens (Ciba Vision Corp., Duluth, GA, USA), has been demonstrated in Figure 1. The MemoryLens is a 3-piece foldable hydrogel that was initially released in 1994.3,4 It has been the most heavily documented IOL with postoperative calcification complications in the United States.6 Several other hydrophilic acrylic lenses have also been implicated in calcific opacification as well (Hydroview, Bausch & Lomb; AquaSense, Ophthalmic Innovations International; SC60B-OUV, Medical Development Research).4,7
Pathophysiology:
The time required from initial lens placement to the development of visually significant opacification in hydrogel lenses can take several years.1,7 The mean interval for the MemoryLens IOL was identified by one study examining 106 explanted lenses to be 25.8 ±11.9 months with a range from approximately 3 months to 6 ½ years.1
MemoryLens predisposition to opacification was believed to be related to the buffering process of the lenses manufactured through the year 1999.1,3,4 The mechanism by which granular calcific opacification (Figure 2) occurs in vivo remains unknown. It is presently believed to be a multifactorial event related to the surface ionization of hydrogel under physiologic pH levels which facilitate calcium precipitation.4,7

Figure 2. High magnification light microscope image demonstrating Alizeran red staining on half of the calcified MemoryLens optic surface compared to an unstained half with granular deposits of calcium.
Risk Factors/Symptoms:
Some of the risk factors that have been associated with hydrogel IOL calcifications include exposure to surgically introduced exogenous substances such as gas, air, tissue plasminogen activator, and silicone oil. Other risk factors include contact with lens packaging materials and lens polishing techniques.1,8 It is currently unknown whether the direct contact of surgical exogenous substances to the optical surface facilitates calcium precipitation or if such sequela is a consequence of increased inflammation resulting from surgical manipulation.7 Progressive visual loss is the most common primary symptom complaint identified in patients with calcified IOLs.6,7
Diagnosis:
Diagnostically, the presence of calcium may be confirmed on pathologic analysis post-explantation by observing characteristic histochemical staining of granules with Alizeran (1,2-dihydroxyanthraquinone) red, as shown in Figure 2 and Figure 3c, and by electron microscopy coupled with energy dispersive x-ray spectroscopy.8 Diffuse granular deposition is typically noted over the lens body, but tends to be more heavily concentrated on the optic center. In the MemoryLens the coated anterior surface shows heavier calcification than the posterior surface which shows less. Early on the posterior surface typically remains free of deposits (Figure 2b and d).3,4 For hydrogel lenses in general, calcium deposition distribution may be superficial, intralenticular, or both.7

Figure 3. Stereotactic images of a calcified MemoryLens following explantation. Lens opacification from anterior views can be appreciated on both unstained (a) and stained (c) lenses with significant granular calcium deposits. A relatively smooth posterior lens surface without deposition is appreciated on views of unstained (b) and stained (d) lens surfaces.
Treatment:
The only treatment currently available for resolving situations of an opacified calcific IOL includes explantation.5
References:
- Werner L. Causes of intraocular lens opacification or discoloration. Cataract Refract Surg. 2007;33:713-726
- Werner L. Biocompatability of intraocular lens materials. Current Opinion in Ophthalmology. 2008;19:41-49
- Haymore J, Zaidman G, Werner L, Mamalis N, Hamilton S, Cook J, Gillette T. Misdiagnosis of hydrophilic acrylic intraocular lens optic opacification: Report of 8 cases with the MemoryLens. Ophthalmology. 2007;114(9):1689-1695
- Neuhann IM, Werner L, Izak AM, Pandey SK, Kleinmann G, Mamalis N, Neuhann TF, Apple DJ. Late postoperative opacification of a hydrophilic acrylic (hydrogel) intraocular lens. Ophthalmology. 2004;111:2094-2101
- Werner L. Calcification of hydrophilic acrylic intraocular lenses. Am J. Ophthalmol. 2008;146(3):341-343
- Mamalis N, Brubaker J, Davis D, Espandar L, Werner L. Complications of foldable intraocular lenses requiring explantation or secondary intervention—2007 survey update. J Cataract Refract Surg. 2008;34:1584-1591
- Gartaganis SP, Prahs P, Lazari ED, Gartaganis PS, Helbig H, Koutsoukos PG. Calcification of hydrophilic acrylic intraocular lenses with a hydrophobic surface: laboratory analysis of 6 cases. Am J Ophthalmol. 2016;168:68-77
- Werner L, Wilbanks G, Ollerton A, Michelson J. Localized calcification of hydrophilic acrylic intraocular lenses in association with intracameral injection of gas. J Cataract Refract Surg. 2012;38:720-721
Bull’s Eye Maculopathy as a Phenotypic Presentation of Stargardt Disease
Home / Retina and Vitreous / Hereditary Retinal and Choroidal Dystrophies
Title: Bull’s Eye Maculopathy as a Phenotypic Presentation of Stargardt Disease
Author: Troy Teeples, MSIV, University of Utah School of Medicine
Photographer: Unknown
Date: 8/14/2017
Image or video:

Image 1: Color fundoscopy shows pigment changes of the macula along with temporal pallor of the optic disc.

Image 2: Fundus Autoflourescence demonstrations central macular hypoautoflourescence with a surrounding hyperautoflourescent ring.
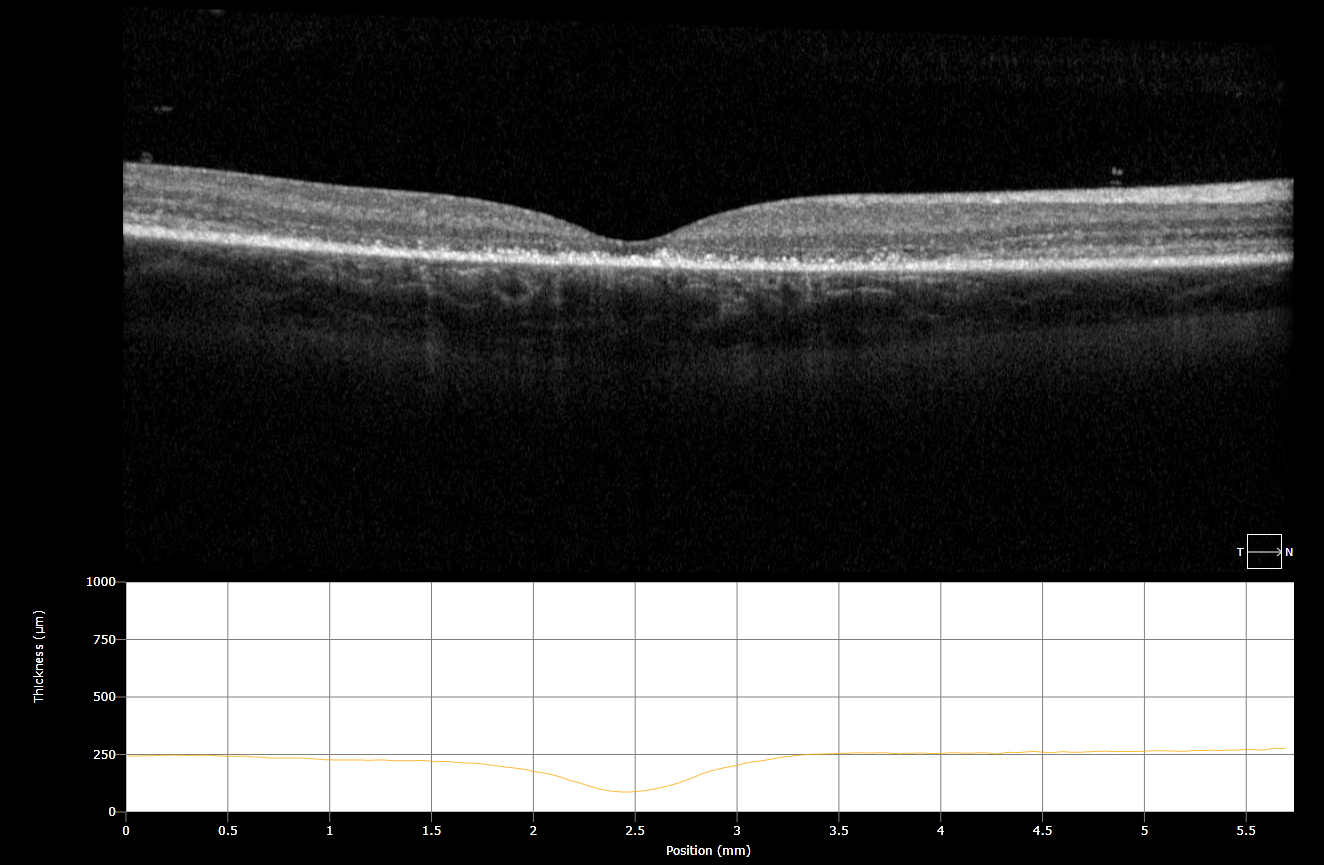
Figure 3: OCT demonstrations a discontinuous ELM layer, IS-OS dropout, and RPE disruption.
Keywords/Main Subjects:
Diagnosis: Stargardt Disease
Description of Image:
Patient Presentation: The patient is a previously healthy 7-year-old boy with no past medical history or family history of ocular disease who originally presented with blurry vision, problems with depth perception and central vision loss. In clinic he was found to have a visual acuity of 20/150 OD, 20/125 OS that was not corrected with refraction along with decreased color vision (Ishihara plates 4/8 OD, 5/8 OS). On fundoscopy he was found to have a Bull’s Eye Maculopathy OU with temporal pallor of the optic disc. No round pisciform flecks were seen on fundoscopy.
Stargardt Disease is the most common inherited macular dystrophy, though it may have many different phenotypic presentations. It is caused by accumulation of lipofuscin in the RPE, resulting from a mutation of the ABCA4 gene. With a faulty ABCA4 gene, the retina is not able to clear a toxic byproduct, bisretinoid-A2E, after the retinoid visual cycle, eventually resulting in RPE atrophy and photoreceptor cell death.
Phenotype Variation: Due to the large size of the ACBA4 gene (6.8kb), there are a significant number of disease-causing variants. In fact, over 500 distinct disease-associated mutations have been discovered that can cause Stargardt disease in addition to other ABCA4 associated diseases such as Cone Dystrophy, AMD, and Retinitis Pigmentosa. This patient presented with a 2 bp deletion resulting in a frameshift mutation on one allele as well as a missense mutation on the other, causing a Bull’s Eye Maculopathy as the phenotypic presentation. While some genotype-phenotype associations have been discovered, many remain yet to be determined. A prospective epidemiological study was performed over 12 months by Cornish and Ho in 2017 in order to determine the incidence of Stargardt Disease in the UK. Phenotypic presentations of the macula were quantified and of the 71 cases used in the study, 4% were found to have no macular abnormalities, 29% were found to have pigment mottling of the macula, 24% had a Bull’s Eye Maculopathy, 37% demonstrated macular atrophy and 36% had round pisciform flecks in the macula.
Imaging: Fundus Autoflouresence revealed a hypoautoflourescent macula with a surrounding hyperautoflourescent ring. An OCT revealed macular thinning, a discontinuous, ‘ratty’ ELM layer, IS-OS dropout, and disruption of the RPE.
References:
- http://eyewiki.aao.org/Stargardt_disease/Fundus_flavimaculatus
- Kurt Spiteri Cornish, Jason Ho, Susan Downes, Neil W. Scott, James Bainbridge, Noemi Lois. The Epidemiology of Stargardt Disease in the United Kingdom. Ophthalmology Retina,Volume 1, Issue 6, 2017, Pages 508-513., ISSN 2468-6530, https://doi.org/10.1016/j.oret.2017.03.001 (http://www.sciencedirect.com/science/article/pii/S2468653017300593)
Faculty Approval by: Griffin Jardine, MD
Footer:
- Copyright statement: Copyright Author Name, ©2016. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
- Attribution/citation suggestions
Disclosure (Financial or other): None
Conjunctival Melanoma
Home / Ophthalmic Pathology / Conjunctiva
Title: Conjunctival Melanoma
Author: Xavier Mortensen, MS4
Photographer: Slit Lamp Photo: Glen Jenkins, CRA, OCTC. Ultrasound Image: Roger Harrie, MD
Date: 6/28/18 (images taken 6/20/18)
Image or video:

Image 1: Slit lamp of left eye showing a 9×9 mm large, vascular, pigmented mass which is non-adherent to the sclera in temporal conjunctiva. Biopsy was consistent with a conjunctival malignant melanoma arising from a preexisting primary acquired melanosis (PAM) with atypia.

Image 2: An immersion scan using a high-frequency 40 MHz probe. The lesion measured 2.7 mm in thickness by 10.3 mm in basal dimension. It appears to only involve the outer 25% of the sclera and with no intraocular extension. The ciliary body also appeared normal without evidence of melanoma.
Keywords/Main Subjects: melanoma, neoplasm, ocular, conjunctiva, sclera, ultrasound, b-scan
Secondary CORE Category: External Disease and Cornea / Neoplasms of the Conjunctiva and Cornea
Diagnosis: Conjunctival Melanoma
Description of Images: see above
Clinical Findings:
Conjunctival melanomas typically present as a nodular brown mass that are often well vascularized. In fact, due to the substantial vascular supply the tumors are more susceptible to bleeding. They occur most commonly on the bulbar conjunctiva or limbus, but may also be found on palpebral conjunctiva. Histologically, isolated or confluent nests of atypical melanocytes are often seen composed of large abnormal cells with high nuclear-cytoplasmic ratio, mitotic figures, and prominent nucleoli. Conjunctival malignant melanomas display invasion into subepithelial layers.
Etiology:
They are more common in patients of European descent and rare in black and Asian populations. Prognosis is poor with an overall mortality rate of 25-45%[3]. Conjunctival melanomas can arise from PAM (70%), nevi (20%), or de novo (10%)[3,4]. Intraocular and orbital extension can occur. It is important to check for an underlying ciliary body melanoma, which can imitate a conjunctival melanoma.
Management:
Treatment is time-sensitive and includes excisional biopsy with cryotherapy and/or alcohol corneal epitheliectomy. Depending on how invasive the malignancy is, exenteration may be required. Ultrasound biomicroscopy (UBM) should be done to rule out extrascleral extension of a ciliary body melanoma of a conjunctival melanoma. The recurrence rate is greater than 50% in treated patients[4], so patients should be followed-up closely by their ophthalmologist.
References:
- Bagheri, Nika, et al. The Wills Eye Manual: Office and Emergency Room Diagnosis and Treatment of Eye Disease. 7th Edition. Philidelphia: Wolters Klewer, 2017.
- Herwig, Martina C. Conjunctival Melanocytic Tumors. 7 November 2017. American Academy of Ophthalmology. 29 June 2018. <http://eyewiki.aao.org/Conjunctival_Melanocytic_Tumors>.
- Kaiser, Peter K, Neil J Friedman and Roberto Pineda. The Massachusetts Eye and Ear Infirmary Illustrated Manual of Ophthalmology. 4th Edition. Elsevier Inc., 2014.
- Skuta, Gregory L, Louis B Cantor and George A Cioffi. “Basic and Clinical Sciences Course.” Section 8: External Disease and Cornea. 2013-2014. American Academy of Ophthalmology, 2013. 227-228.
Faculty Approval by: Roger Harrie, MD; Griffin Jardine, MD
Footer:
- Copyright statement: Copyright Author Name, ©2016. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
- Attribution/citation suggestions
Disclosure (Financial or other): None
Horner’s Syndrome in a 17 Year Old
Home / Neuro-Ophthalmology / Systemic Conditions with Neuro-Ophthalmic Signs
Title: Horner’s Syndrome in a 17 year old
Author: Emily Ross, 4th Year Medical Student at Indiana University-Purdue University Indianapolis
Date: 9/21/2017
Keywords/Main Subjects: Horner’s syndrome, anisocoria, ptosis
CORE Category:
Neuro-Ophthalmology > Pupillary Abnormalities > Anisocoria
Diagnosis: Horner’s Syndrome
Description of Case:
The patient is a 17-year-old girl who presented with ptosis on the right and anisocoria that began four months prior to presentation at clinic. She denies any cough, chest pain, neck pain, eye pain, neck manipulation, trauma, or preceding illnesses. She has no other significant findings on exam and no significant medical history. She does not use any eye drops. Differential diagnosis: Horner’s Syndrome, physiologic anisocoria, Adie tonic pupil, or topical/systemic medications. The patient was found to have Horner’s Syndrome.
Horner’s Syndrome:
Horner’s syndrome can result from a lesion anywhere along the three neuron oculosympathetic pathway. It can produce three main deficits: ipsilateral ptosis (eyelid drooping) because of loss of innervation to Muller’s muscle, ipsilateral miosis (pupillary constriction) because of unopposed parasympathetic drive to the pupil, and ipsilateral anhydrosis (lack of sweating) on the face that may be best appreciated by the patient as contralateral hemifacial redness during exertion. This hemifacial redness is called the harlequin sign.
When evaluating a patient with anisocoria, first check that both pupils react appropriately to light, then determine if the difference in pupil size is greater in dim light or bright light. Horner’s Syndrome will have a greater difference seen between the two pupils in dim light. Dilation lag will also be present in Horner’s Syndrome and can help differentiate it from physiologic anisocoria. When the normal pupil dilates, there is both a parasympathetic signal allowing relaxation of the sphincter and a sympathetic signal contracting the pupillary dilator muscle. In Horner’s syndrome, the sympathetic signal is absent so the pupil only dilates by parasympathetic sphincter relaxation which is not as brisk resulting in a slower dilation response to dim light.

Figure 1: Dilation lag: After five seconds in dim light, the left (unaffected) pupil has dilated to 7mm, while the right (affected) pupil is only dilated to 5mm. After 15 seconds, the left pupil is dilated to 7.5mm and the right is dilated to almost 7mm.
Confirming the diagnosis of Horner’s Syndrome can be accomplished pharmacologically using either cocaine or aproclonidine eye drops. Cocaine is a norepinephrine reuptake inhibitor. When instilled in a patient with Horner’s Syndrome, the unaffected pupil will dilate significantly while the affected pupil will have a minimal or no response. The sympathetic innervation to the affected pupil is absent so the cocaine will have no (or very little) norepinephrine present in the synapse to prevent reuptake of resulting in little or no dilation of the pupil. Additionally, after a positive cocaine test, one can apply topical phenylephrine to the miotic pupil which directly stimulates sympathetic receptors resulting in dilation of the affected pupil. Aproclonidine is a strong alpha-2 adrenergic agonist and a weak alpha-1 adrenergic agonist. In the unaffected pupil, the alpha-2 stimulation results in pupillary constriction but in the affected pupil, the alpha-1 receptors become hypersensitive about one week after the sympathetic denervation and will cause pupillary dilation reversing the anisocoria as well as improvement of the ptosis.
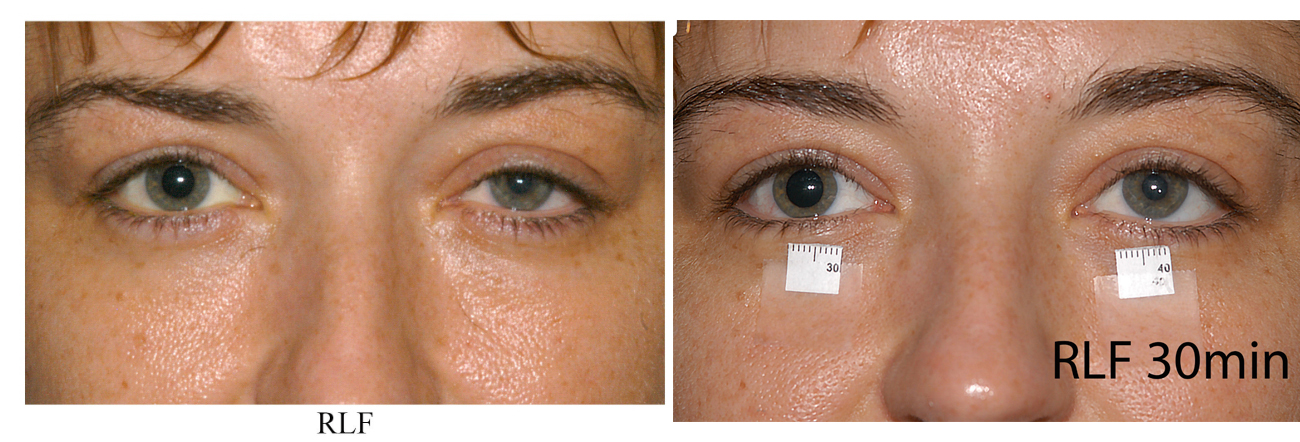
Figure 2: Cocaine eye drop test: Upon presenting to clinic, the patient shows anisocoria with a smaller left pupil and ptosis on the left. Thirty minutes after application of 4% cocaine eye drops in room light, the right pupil has dilated significantly while the left pupil remains unaffected confirming the diagnosis of Horner’s Syndrome.
Localizing the lesion to either a first order or second order neuron lesion (preganglionic) versus a third order neuron (postganglionic) lesion can be accomplished pharmacologically by using hydroxyamphetamine eye drops though these drops are not routinely available even at large institutions. If the postganglionic neuron is functioning, the hydroxyamphetamine will cause release of norepinephrine from the neuron resulting in pupil dilation and narrowing the differential to a lesion in either the central or preganglionic neurons. If there is no dilation, then the lesion has been localized to the postganglionic neuron. If a cocaine test is performed, one must wait at least 24 hours before performing the hydroxyamphetamine test as the cocaine can interfere with this localizing test. Additionally, history and exam findings may also assist the clinician in localizing the lesion.
Understanding the anatomy of the oculosympathetic pathway is crucial. First order or central neurons run from the hypothalamus through the brainstem and spinal cord to the level of C8/T1. Second order or preganglionic neurons exit the brainstem and ascend to the superior cervical ganglion located near the carotid artery bifurcation. On this route, second order neurons pass near the subclavian artery and lung apex. Third order or postganglionic neurons begin at the superior cervical ganglion and run with the internal carotid artery.
Imaging of the head, neck, and chest may be required to establish an underlying cause for the Horner’s syndrome with either MRI, CT, angiography, or ultrasound. Wallenberg Syndrome, carotid artery dissections, Pancoast or apical lung tumors, basal skull tumors, dislocation of cervical vertebrae, aortic dissection or aneurysm, and iatrogenic causes are a few potential etiologies of a Horner’s Syndrome to investigate.
References:
- Ophthalmology AA of. 2017-2018 Basic and Clinical Science Course Neuro-Ophthalmology. S.I.: American Academy of Ophthalmology; 2017.
- Kanagalingan S, Miller NR. Horner syndrome: clinical perspectives. Eye and Brain. April 2015; (10)7:35-46. doi: 10.2147/EB.S63633.
- Davagnanam I, Fraser CL, Miszkiel K, Daniel CS, Plant GT. Adult Horner’s syndrome: a combined clinical, pharmacological, and imaging algorithm. Eye. March 2013; 27(3):291-8. doi: 10.1038/eye.2012.281.
- Kedar S, Biousse V, Newman N. Horner Syndrome. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. http://www.uptodate.com (Accessed on September 23, 2017.)
Identifier: Moran_CORE_24942
Copyright statement: Copyright 2018. Please see terms of use page for more information.
Two Cases of Wolfram Syndrome
Home / Neuro-Ophthalmology / Grand Rounds Presentations and Cases
Title: Two Cases of Wolfram Syndrome
Author: Nathan G. Lambert, BS; Kathleen B. Digre, MD
Keywords: Wolfram Syndrome; WFS1; Wolframin; Diabetes insipidus; Diabetes mellitus; Optic atrophy; Sensorineural deafness;
Secondary CORE Location: Neuro-Ophthalmology / Systemic Conditions with Neuro-ophthalmic Signs
Diagnosis: Wolfram Syndrome
Intro
Wolfram Syndrome, or DIDMOAD, is a rare genetic syndrome consisting of diabetes insipidus, diabetes mellitus, optic atrophy and deafness. It was first reported in 1938 by Wolfram and Wagener who reported a family of 8 siblings, 4 of whom had juvenile diabetes mellitus and optic atrophy.1 We report two cases of siblings who exhibited typical findings of Wolfram Syndrome and describe their disease course and progression.
Case 1
A 10-year-old boy with a family history of Wolfram Syndrome presented to Moran neuro-ophthalmology clinic after referral for possible hereditary optic atrophy. The patient had a 5-year preceding history of diabetes mellitus and high frequency hearing loss. During elementary school he experienced increased urinary frequency and was eventually diagnosed with diabetes insipidus and treated with DDAVP. His older brother had also recently tested positive for Wolfram Syndrome. His vision was 20/30 OD and 20/20 OS with mild correction. Color vision was 5/6 OD and 6/6 OS, with full stereo 9/9. No APD was noted at the time.
At age 12 later he was noted to have some slight temporal pallor. His color vision was 6/7 with stereoscopic vision of 8/9. At age 14, his genetic testing returned positive for a mutation in the Wolfram gene (WFS1), located at map position 4p16.1. He had also experienced some decrease in hearing. He continued to require insulin therapy for his diabetes mellitus.
Between the age of 16 to 18, he was noted to have some peripapillary atrophy on funduscopic examination, and Humphrey visual field (HVF) testing showed an enlarged blind spot with mean deviation of -4.65 (OD) and -5.07 (OS) (Figure 1). His pupils were noted to be slightly sluggish to light reaction with a slight near dissociation. His color vision had decreased to 7/9 but his stereoscopic vision remained full.
At his most recent appointment, at age 22, his vision continued to be stable, but hearing loss in right ear appeared to be worse than previously. His stereoscopic and color vision were also decreased at 4/9 (stereo) and 1/10 (OD) and 2/10 (OS) (color). OCT revealed thinning of the retinal nerve fiber layer (RNFL) He still showed no APD at this time, however his pupils continued to react sluggishly to light. At this point he had started to report swallowing difficulties.
Case 2
This the older brother of patient in Case 1 was sdiagnosed with diabetes mellitus at age 7. He was later diagnosed with diabetes insipidus and treated with DDAVP. At age 10 he failed the school eye exam and presented to neuro-ophthalmology clinic where he was found to have decreased color vision and a VA (CC) of 20/200 (OD) and <20/400 (OS). He scored 0/7 for color vision and 6/9 for stereoscopic vision. His pupils were very poorly reactive but showed no afferent pupillary defect (APD). Funduscopic exam showed a pale nerve and OCT showed extensive retinal nerve fiber layer loss. His visual fields showed an enlarged blind spot bilaterally. Later that year an MRI showed atrophy and thinning of the brainstem with cerebellar hemispheric atrophy.
Throughout this process he was found to have depression, anxiety, and obsessive-compulsive disorder (OCD) and had previously been diagnosed with Asperger’s Syndrome. Genetic results at age 14 showed two mutations within exon 8 of the WFS1 gene and he was officially diagnosed with Wolfram Syndrome.
His condition continued to decline over the next 8 years. By age 22 he was placed on a CPAP for obstructive sleep apnea (OSA). By age 23 he exhibited swallowing difficulties and presented to the emergency department on multiple occasions for dysphagia and frequent choking episodes. He had also started to develop an uncoordinated, wide-based gait. At age 25 he experienced another episode of aspiration whereby he eventually died of pneumonia and respiratory failure.
Discussion
Wolfram Syndrome is neurodegenerative condition due to a loss of function mutation in the WFS1 gene, which codes for the protein wolframin1. This rare genetic disease is thought to occur in 1:770,0002, with an average life expectancy of 40 years3. Although genetic testing is necessary for definitive diagnosis, Wolfram Syndrome should be suspected in any patients with an inherited association of juvenile-onset (before age 16) insulin-dependent diabetes mellitus and progressive bilateral optic atrophy4.
Genetics
The WFS1 gene codes for the protein wolframin, and endoglycosidase H-sensitive membrane glycoprotein that localizes primarily to the endoplasmic reticulum (ER)5. This protein is thought to function as an ER calcium channel or regulator of calcium channel activity, and as such is important in cell-to-cell communication, muscle contraction, and protein processing6. Fonseca et al. found that WFS1 was upregulated during glucose-induced insulin secretion. They also noted that knockdown of WFS1 resulted in an ER stress signal, leading to beta-cell dysfunction and cell death, and subsequent diabetes7. Deletion or loss-of-function mutations in WFS1 result in ER dysfunction, leading to apoptosis of that associated cell.
Diabetes Insipidus
Seventy percent of patients with Wolfram Syndrome go on to develop central diabetes insipidus1, due to loss of vasopressin producing neurons. Proper functioning of the WFS1 gene is important for maintenance of neuron’s responsible for vasopressin synthesis and processing. Gabreels et al. found that patients with Wolfram Syndrome had vasopressin neuron loss in the supraoptic nucleus as well as defects in vasopressin precursor proteins8.
Diabetes Mellitus
Impaired glucose control is usually one of the first signs of Wolfram Syndrome. Diabetes mellitus is often diagnosed by age 61, Wolframin is highly expressed in the pancreatic beta-cells and may assist in folding mechanisms of insulin precursor proteins7. WFS1 deficient mice have destruction of beta-cells resulting in impaired glucose tolerance and diabetes mellitus9. Patients with Wolfram Syndrome often remain insulin dependent throughout the course of their life.
Optic Atrophy
Optic atrophy, noticed as loss of color and peripheral vision, is usually the second symptom behind diabetes mellitus, often occurring by age 111. Optic atrophy occurs in almost all patients10, and most patients eventually go blind1. Wolframin is localized to retinal ganglion cells, inner nuclear layer photoreceptors and glial cells of the retina11. The complete cellular mechanism resulting in optic atrophy is unclear, but is thought to be from issues in proper ER function leading to protein deficits, axonal transport deficiencies, and subsequent optic atrophy1.
Deafness
The mechanistic etiology of sensorineuronal hearing loss seen from WFS1 mutations has also not been fully elucidated. Some studies suggest that proper wolframin function is necessary for maintenance of cochlear hair cells12. It has also been suggested that calcium dysregulation that occurs secondary to WFS1 mutations is implicated in resultant deafness12. It is likely that both mechanisms are at work, however further study is needed to clarify this process.
Mental Illness
Neurologic and psychological complications are also common in patients with Wolfram Syndrome. Takeda et al found that WFS1 mRNA and protein were highly expressed in the amygdala, hippocampus, and other areas of the limbic system13. Patients with Wolframin Syndrome are often affected with severe mental and emotional illness such as anxiety, depression, psychosis, and aggression14. WFS1 gene is also expressed in the raphe nucleus and nucleus ceruleus, making it logical that mutations leading to imbalanced levels of serotonin and norepinephrine could lead to impulsive suicide and psychiatric disease15. One study looking at MRI findings in Wolfram Syndrome patients found associated generalized brain atrophy, especially in the cerebellum, medulla, and pons16. Interestingly, one of the most common causes of death in these patients is central respiratory failure secondary to severe brainstem atrophy17,18.
Other Manifestations
A variety of other pathologic associations have been seen occur commonly in patients with Wolfram Syndrome. By their early 20’s, many patients with WS experience incontinence1 secondary to neurogenic bladder and other urinary tract or bladder abnormalities19. Many patients develop cerebellar ataxia resulting in gait and balance issues10. Other issues include peripheral neuropathy, loss of gag reflex, myoclonus, mental retardation, seizures, and dementia1.
Conclusion
The two patient cases presented exhibit a variety of common findings of Wolfram Syndrome including, diabetes insipidus, diabetes mellitus, progressive optic atrophy, sensorineural deafness, swallowing difficulties, mental illness (anxiety, depression), bladder incontinence, cerebellar ataxia, and death secondary to aspiration and central respiratory failure. Any patient with a personal and family history of juvenile onset diabetes mellitus and bilateral progressive optic atrophy should be suspected and evaluated for Wolframin Syndrome.
Bibliography
- Rigoli, L., Lombardo, F. & Di Bella, C. Wolfram syndrome and WFS1 gene. Clin Genet 79, 103-117 (2011).
- Ganie, M.A. & Bhat, D. Current developments in Wolfram syndrome. J Pediatr Endocrinol Metab 22, 3-10 (2009).
- Kinsley, B.T., Swift, M., Dumont, R.H. & Swift, R.G. Morbidity and mortality in the Wolfram syndrome. Diabetes Care 18, 1566-1570 (1995).
- Khanim, F., Kirk, J., Latif, F. & Barrett, T.G. WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Hum Mutat 17, 357-367 (2001).
- Garcia, J.B., Venturino, M.C., Devesa, G. & Basabe, J.C. Insulin secretion induced by alloantigens. Mechanisms of action. Acta Diabetol Lat 26, 283-289 (1989).
- Osman, A.A., et al. Wolframin expression induces novel ion channel activity in endoplasmic reticulum membranes and increases intracellular calcium. J Biol Chem 278, 52755-52762 (2003).
- Fonseca, S.G., et al. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem 280, 39609-39615 (2005).
- Gabreels, B.A., et al. The vasopressin precursor is not processed in the hypothalamus of Wolfram syndrome patients with diabetes insipidus: evidence for the involvement of PC2 and 7B2. J Clin Endocrinol Metab 83, 4026-4033 (1998).
- Ishihara, H., et al. Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet 13, 1159-1170 (2004).
- Tranebjaerg, L., Barrett, T. & Rendtorff, N.D. WFS1-Related Disorders. (1993).
- Yamamoto, H., et al. Wolfram syndrome 1 (WFS1) protein expression in retinal ganglion cells and optic nerve glia of the cynomolgus monkey. Exp Eye Res 83, 1303-1306 (2006).
- Cryns, K., et al. Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum Mutat 22, 275-287 (2003).
- Takeda, K., et al. WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 10, 477-484 (2001).
- Swift, M. & Swift, R.G. Wolframin mutations and hospitalization for psychiatric illness. Mol Psychiatry 10, 799-803 (2005).
- Sequeira, A., et al. Wolfram syndrome and suicide: Evidence for a role of WFS1 in suicidal and impulsive behavior. Am J Med Genet B Neuropsychiatr Genet 119B, 108-113 (2003).
- Hardy, C., et al. Clinical and molecular genetic analysis of 19 Wolfram syndrome kindreds demonstrating a wide spectrum of mutations in WFS1. Am J Hum Genet 65, 1279-1290 (1999).
- Sam, W., Qin, H., Crawford, B., Yue, D. & Yu, S. Homozygosity for a 4-bp deletion in a patient with Wolfram syndrome suggesting possible phenotype and genotype correlation. Clin Genet 59, 136-138 (2001).
- Barrett, T.G., Bundey, S.E. & Macleod, A.F. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 346, 1458-1463 (1995).
- Cremers, C.W., Wijdeveld, P.G. & Pinckers, A.J. Juvenile diabetes mellitus, optic atrophy, hearing loss, diabetes insipidus, atonia of the urinary tract and bladder, and other abnormalities (Wolfram syndrome). A review of 88 cases from the literature with personal observations on 3 new patients. Acta Paediatr Scand Suppl, 1-16 (1977).
Legend

Humphrey visual field (HVF) showing progression of enlarging blind spot.

Humphrey visual field (HVF) showing progression of enlarging blind spot.
Identifier: Moran_CORE_24609
39-year-old Male with New-Onset Right Vision Loss
Home / Retina and Vitreous / Focal and Diffuse Choroidal and Retinal Inflammation
Title: 39-year-old male with new-onset right vision loss
Author: Nina Boal, BA
Photographer: James Gilman, CRA, FOPS
Date: September 2017
Images:

Figure 1: Dilated fundus examination of the right eye on presentation

Figure 2: MRI of orbits with IV contrast, post-contrast T1 5 mm ring enhancing foci in the right thalamus.

Figure 3: Dilated fundus examination of the right eye after 1.5 months of treatment
Keywords/Main Subjects: Toxoplasma gondii; Toxoplasma retinitis; HIV; AIDS
Secondary CORE Category: Intraocular Inflammation and Uveitis / Ocular Involvement in AIDS (HIV)
Diagnosis: Toxoplasma Retinitis
Discussion of Images:
The fundus photos and MRI are from a 39-year-old male who presented with right sided vision loss over the course of two days. A complete review of systems was positive only for ten-pound weight loss in three months, fatigue, nausea, and bloody stools. The patient immigrated from Guatemala 25 years ago. He endorsed no significant past medical history.
- Vision: OD 20/70; OS 20/20
- Intraocular pressure: normal OU
- Slit lamp examination OU: unremarkable
- Dilated Exam:
- OS unremarkable
- OD elevated, white sub-retinal lesion in the inferior arcade with overlying vitreous inflammatory haze and hemorrhage was seen in the right eye (Figure 1)
The patient was treated empirically with intravitreous clindamycin and foscarnet, intravenous acyclovir, oral trimethoprim/sulfamethoxazole and admitted for work up of endogenous endophthalmitis. Further testing revealed:
- Pancytopenia with a white blood cell count of 2.4
- HIV testing positive; CD4+ T-cell count of 24 cells/mm3; viral load of 1,400,000
- MRI with IV contrast:
- 5 mm ring enhancing foci in the right thalamus (figure 2), in addition to small enhancing lesions in the anterior right frontal lobe and right basal ganglia
- Imaging was read as concerning for neurocystercercosis or toxoplasma
- Vitreous sampling: Toxoplasma gondii PCR positive and CMV negative
The patient was diagnosed with toxoplasma retinitis and AIDS. Other medications started after this admission were emtiracitabine/tenofovir alafenamide and raltegravir. Valgancyclovir and azithromycin were started for CMV and MAC prophylaxis.
Follow-up appointment 1.5 months after treatment began showed improved vision to 20/40 in the right eye. Dilated fundus exam of the right eye showed improved subretinal fluid and hemorrhages with slightly larger area of retinal whitening along the inferior arcade (Figure 3).
Discussion:
Ocular toxoplasmosis, caused by the protozoan parasite Toxoplasma gondii, is considered to be the most common identifiable cause of infectious posterior uveitis in many parts of the world including northern Europe, North America, and South America. The immune system plays a critical role in susceptibility to infection with toxoplasmosis, and patients are at a particularly high risk when their CD4+ T-cell count is below 200 cells/mm3.
Active ocular toxoplasmosis can appear as focal white fluffy retinal lesions with indistinct borders. Overlying vitreous inflammation is commonly seen, with more severe overlying vitreous inflammation representing more advanced active disease, and classically described as a “headlight in a fog.” Older ocular toxoplasmosis lesions are typically seen as areas of well-circumscribed retinal necrosis, and as the lesion heals, its borders become more defined and hyperpigmented. Ocular toxoplasmosis in an immunocompromised patient typically demonstrates a more fulminant course with multifocal disease in one eye, bilateral eye disease, or extensive areas of necrotizing retinitis that can progress to panophthalmitis and orbital cellulitis. Between 30 and 50 percent of patients with HIV who have ocular toxoplasmosis will have central nervous system involvement.
In healthy patients with peripheral retinal lesions, infection with T. gondii is self-limited and may not require treatment, however treatment is required for patients who are immunocompromised, pregnant, or have vision threatening lesions. There have been three prospective, randomized, placebo-controlled clinical trials using systemic antibiotics to treat ocular toxoplasmosis. However, two of these studies were conducted almost 40 years ago and they are all considered to be methodologically poor. In the three studies, there was a lack of evidence that antibiotics (short or long term) would prevent vision loss. The “classical triple therapy” remains the combination of oral pyrimethamine, sulfadiazine, and corticosteroids. Trimethoprim/sulfamethoxazole is required for neurotoxoplasmosis, as in this case, and intravitreous clindamycin in combination with dexamethasone was shown to be efficacious in 16 out of 16 patients in a recent study (Zamora YF et al., 2015). Caution may be warranted in the use of steroids for immunocompromised patients.
Format: Images
References:
- Cunningham ET Jr, Margolis TP. Ocular manifestations of HIV infection. N Engl J Med 1998; 339: 236–44.
- Holland GN. Ocular toxoplasmosis: a global reassessment. Part I: epidemiology and course of disease. Am J Ophthalmol 2003; 136:973-88.
- Holland GN. Ocular toxoplasmosis: a global reassessment. Part II: disease manifestations and management. Am J Ophthalmol 2004; 137:1-17.
- Maenz M, Schluter D, Liesenfeld O, Schares G, Gross U, Pleyer U. Ocular toxoplasmosis past, present and new aspects of an old disease. Progress in Retinal and Eye Research 2014; 38: 77-106.
- Moshfeghi DM, Dodds EM, Couto CA et al. Diagnostic approaches to severe, atypical toxoplasmosis mimicking acute retinal necrosis. Ophthalmology 2004; 111: 716–25.
- Talabani H, Mergey T, Yera H, Delair E, Brezin AP, Langsley G, Dupouy-Camet J. Factors of occurrence of ocular toxoplasmosis. Parasite 2010; 17: 177-182.
- Zamora YF, Arantes T, Reiz FA, Garcia CR, Saraceno JJ, Belfort JR, Muccioli C. Local treatment of toxoplasmic retinochoroiditis with intravitreal clindamycin and dexamethasone. Arg Bras Oftalmol 2015; 78 (4): 216-9.
Faculty Approval by: Griffin Jardine, MD
Copyright: Nina Boal, © For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
Disclosure (Financial or other): None
Identifier: Moran_CORE_24588