


Coats Disease Masquerading as Panuveitis
Home / Retina and Vitreous / Other Retinal Vascular Diseases
Title: Coats Disease Masquerading as Panuveitis
Authors: Christopher Le, MSIV University of Colorado School of Medicine; Eric Hansen, MD.
Date: 7/8/2023
Keywords/Main Subjects: coats disease, uveitis masquerade syndromes
Diagnosis: Coats Disease
Description of Case:
Introduction:
Coats disease is a rare, congenital, nonhereditary, idiopathic disorder of the retinal vasculature leading to massive subretinal and intraretinal exudation. The disease typically affects young males in the first or second decades of life and is unilateral.1 The pathophysiology of Coats disease remains unclear – however, it has been proposed that defects in endothelial cells of the retinal vasculature along with abnormal pericytes contribute to the breakdown of the blood-retinal barrier leading to the development of telangiectatic vessels, retinal ischemia, and exudation of lipid-rich fluid.2 Patients with Coats disease have varying symptoms on initial presentation including: decreased visual acuity, eye pain, strabismus, and leukocoria.3 Rarely, Coats disease may present with symptoms and exam findings suggestive of intraocular inflammatory conditions such as uveitis or endophthalmitis. Diseases that present with an intraocular inflammatory picture but are not caused by immune-mediated or infectious processes are referred to as uveitis masquerade syndromes (UMS).4
Case Presentation:
We present a case of Coats disease in a 14-year-old male masquerading as a panuveitis. The patient presented to the emergency department at an outside institution with a three-day history of blurred vision, pain, and redness in his left eye, as well as fever, body aches, and a facial rash. On initial examination, visual acuity in his left eye was markedly decreased at light perception only. There was conjunctival injection and diffuse cell in the anterior chamber and vitreous. On dilated funduscopic examination, a multifocal exudative retinal detachment without macular involvement was appreciated with diffuse subretinal and intraretinal yellow-white exudation and peripheral retinal hemorrhages. Interestingly, the patient reported having a normal dilated eye exam just one year prior.
Shortly after presentation to the outside facility, the patient was transferred to the Moran Eye Center at the University of Utah. Due to concern for possible endophthalmitis or infectious panuveitis, the patient underwent a vitreous and anterior chamber tap as well as injection of broad-spectrum antimicrobials. The patient was evaluated by uveitis specialist who recommended a broad uveitis workup and vitreous biopsy given a dry vitreous tap. The patient’s respiratory viral panel resulted positive for rhinovirus. Broad uveitis workup – including evaluation for HSV, VZV, CMV, toxoplasma, vitreous PCR, quantiferon, RPR, syphilis antibody, bartonella, lysozyme, ACE, and ANCA – returned unremarkable. The patient underwent an exam under anesthesia and a 27-gauge pars plana vitrectomy with vitreous biopsy and intravitreal injections of antimicrobial agents including vancomycin, ceftazidime, clindamycin, and foscarnet. Interestingly, the culture from patient’s vitreous biopsy grew pan-sensitive staphylococcus Capitis. However, the patient’s Karius panel was negative for bacteria and positive for candida. Both positive results were determined to be likely contaminants in consultation with the infectious disease department.
The patient followed-up in clinic and underwent fundus photography and fluorescein angiography following vitrectomy. Fundus examination demonstrated telangiectatic vessels and bulbs with associated diffuse subretinal exudative material and exudative retinal detachment. Fluorescein angiography highlighted the telangiectatic vessels and demonstrated terminal light bulb aneurysms, capillary nonperfusion, and perivascular leakage. The patient was diagnosed with Coats disease, stage 3A given this constellation of findings. The patient was scheduled to undergo laser photocoagulation therapy with adjuvant intravitreal therapy.
At the time of treatment, he was found to have diffuse vitreous hemorrhage obscuring a clear view of the retina with worsening exudative retinal detachment. Subsequently, he was taken to the operating room for a pars plana vitrectomy with scleral buckling, external drainage of exudative material, application of endolaser, silicone oil endotamponade, and intravitreal Avastin injection. At the time of this vitrectomy, the patient’s exudative retinal detachment had worsened to total detachment –classifying the patient’s disease as stage 3B. The patient subsequently underwent multiple exams under anesthesia with laser photocoagulation, sub-tenons Kenalog and intravitreal Avastin injections resulting in resolution of the exudative retinal detachment, significantly improved exudation, and residual fibrosis and vitreoretinal traction sparing the macula. The patient’s visual acuity has improved to counting fingers, and he remains without any signs of neovascular glaucoma.
Discussion:
This case highlights a rare and atypical presentation of Coats disease masquerading as a panuveitis. The critical step in obtaining this patient’s diagnosis, and in diagnosis of Coats disease in general, was obtaining fluorescein angiography with clear view which demonstrated pathognomonic features of the disease: telangiectatic retinal vasculature with terminal light bulb aneurysms, capillary nonperfusion, and perivascular leakage. Coats disease should be included in the differential diagnosis for young males in the first or second decades of life presenting with panuveitis and exudative retinal detachments.
Images or video:
Image 1

Image 1: Fundus photograph taken after initial vitrectomy demonstrating telangiectatic retinal vasculature, yellow subretinal exudative material, and multifocal exudative retinal detachment.
Image 2
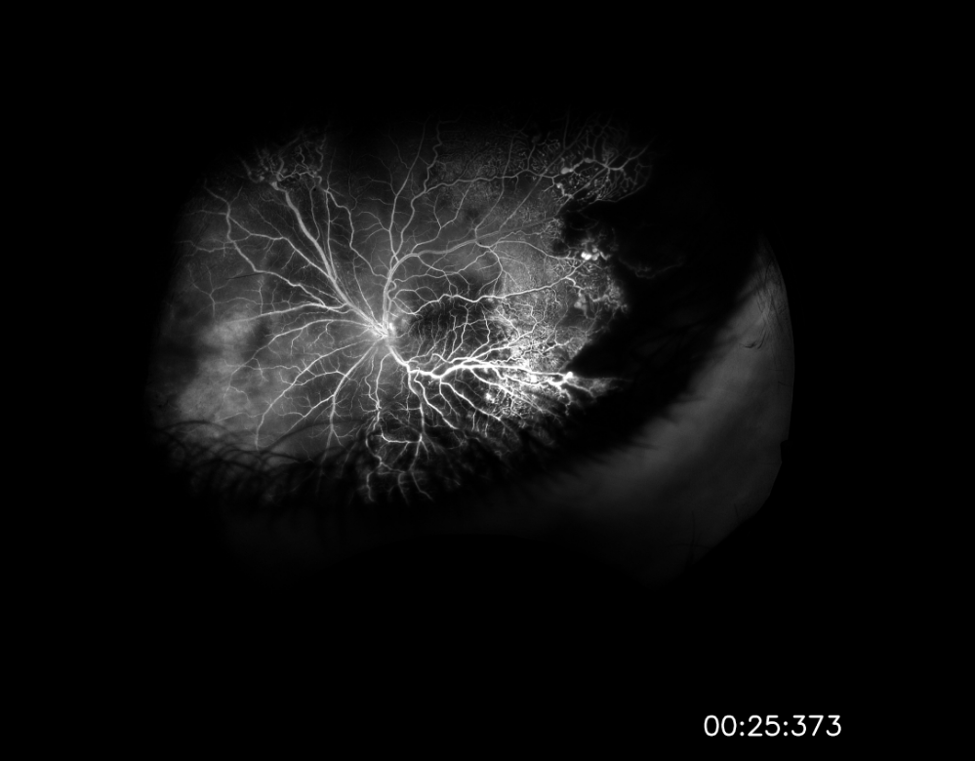
Image 2: Widefield fluorescein angiography taken after initial vitrectomy demonstrating telangiectatic vessels with terminal light bulb aneurysms, capillary nonperfusion, and perivascular leakage.
Image 3

Image 3: Fundus photograph taken during most recent exam under anesthesia in June 2023, following several laser photocoagulation and intravitreal Avastin treatments, demonstrating improved exudative retinal detachment, persistent but improved exudation, and residual fibrosis and traction involving the macula.
Summary of the Case:
A 14-year-old male presented with a marked decrease in visual acuity to light perception only, eye pain, and eye redness of the left eye in the setting of fever, chills, and a facial rash. Initial examination was remarkable for a multifocal exudative retinal detachment and significant anterior chamber and vitreous cell suggestive of a panuveitis or intraocular inflammatory condition. Vitreous culture from intraoperative biopsy grew staphylococcus Capitis, but was ultimately determined to be a contaminant. All other elements of the patient’s broad uveitis workup were unremarkable. Fluorescein angiography was performed following the patient’s initial vitrectomy and revealed telangiectatic vessels, capillary nonperfusion, perivascular leakage – findings pathognomonic for Coats disease. The patient underwent a series of interventions including vitrectomy/buckle with external drainage of exudates and several treatments with laser photocoagulation and intravitreal Avastin injections. His most recent examination and fundus photos demonstrated improvement in the exudative retinal detachment and persistent, but improved, exudation. His visual acuity has improved to counting fingers and he remains without any signs of development of neovascular glaucoma.
Format: Case Report
References
-
- Yousef YA, ElRimawi AH, Nazzal RM, et al. Coats’ disease: characteristics, management, outcome, and scleral external drainage with anterior chamber maintainer for stage 3b disease. Medicine (United States). 2020;99(16):E19623. doi:10.1097/MD.0000000000019623
- Sen M, Shields CL, Honavar SG, Shields JA. Coats disease: An overview of classification, management and outcomes. BMC Ophthalmol. 2017;17(1):1. doi:10.4103/ijo.IJO
- Yang X, Wang C, Su G. Recent advances in the diagnosis and treatment of Coats’ disease. Int Ophthalmol. 2019;39(4):957-970. doi:10.1007/s10792-019-01095-8
- Hsu YR, Wang LU, Chen FT, et al. Clinical Manifestations and Implications of Nonneoplastic Uveitis Masquerade Syndrome. Am J Ophthalmol. 2022;238(August 2020):75-85. doi:10.1016/j.ajo.2021.12.018
Faculty Approval by: Eric Hansen, MD; Griffin Jardine, MD.
Copyright: Christopher Le, © 2023. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Identifier: Moran_CORE_126934
Enhanced S-cone Syndrome: an NR2E3-Associated Retinal Dystrophy
Home / Retina and Vitreous / Hereditary and Choroidal Dystrophies
Title: Enhanced S-cone Syndrome: an NR2E3-Associated Retinal Dystrophy
Authors: Aniket Ramshekar, PhD, MSIV, University of Utah School of Medicine and Cecinio “Nikko” Ronquillo, MD, PhD
Date: 8/23/23
Keywords/Main Subjects: enhanced s-cone syndrome, NR2E3, NRL, nyctalopia, photoreceptors
Diagnosis: Enhanced S-cone Syndrome
Description of Case: A 13-year-old male presented with impaired nighttime vision (i.e., nyctalopia) and peripheral vision loss. Past ocular history is notable for glasses since he was 4 years old. Medical and developmental history were unremarkable. Family history was unremarkable.
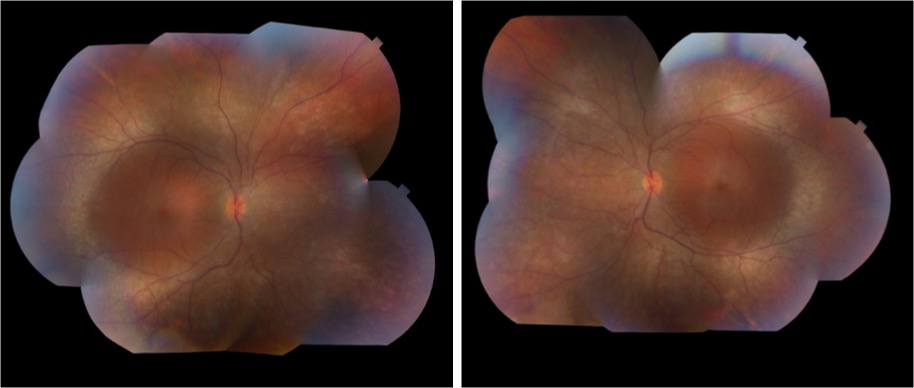
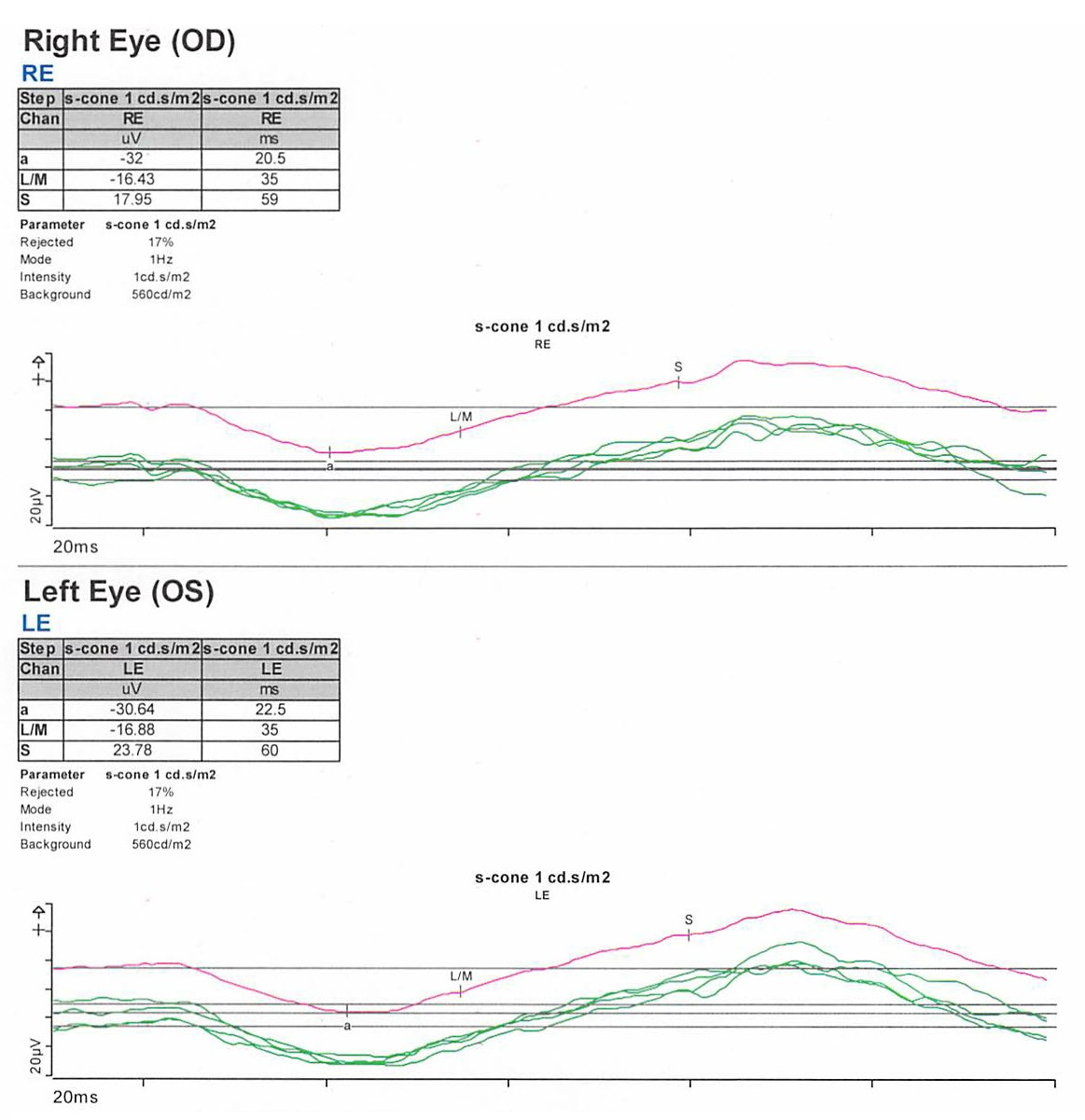
On exam, the patient had best corrected visual acuity (BCVA) of 20/30 OD and 20/40 OS, intraocular pressure (IOP) of 12 OD and 9 OS, and reduced peripheral vision to confrontation OU. Fundus exam was notable for loss of pigment, grayish appearance around the arcades and peripheral retina associated with nummular pigment changes along the superior arcade OS (Figure 1).
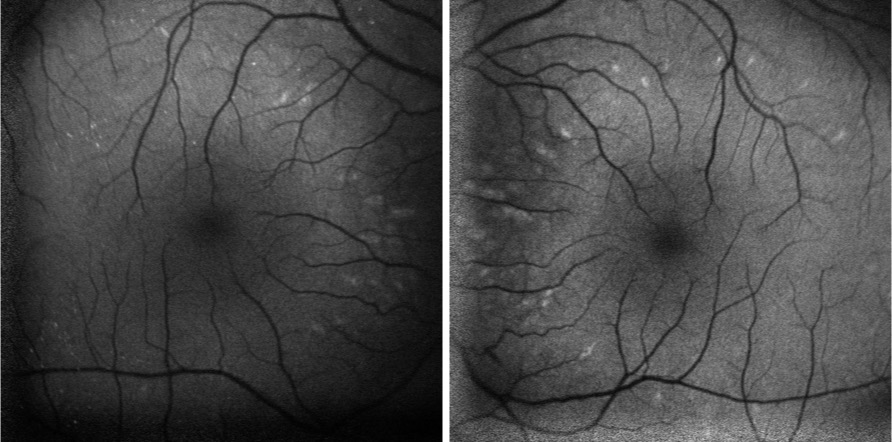
Fundus autofluorescence revealed hyper-autofluorescent spots surrounding the fovea OU (Figure 2).
Optical coherence tomography (OCT) demonstrated retained inner layers of the retina OU, cystic changes in the outer plexiform layer (OPL) OU, and loss of definition of the outer retinal layers OU (Figure 3).

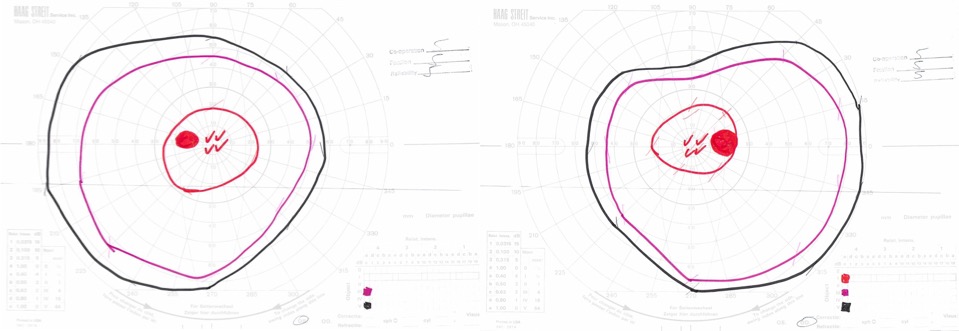
Goldmann visual field exam was full (Figure 4).
Full-field electroretinogram (ERG) showed diminished scotopic and photopic bright white flashes OU and 30 Hz flicker response that was delayed and diminished in amplitude OU (Figure 5).

Given the concern for inherited retinal dystrophy, the patient was referred for genetic testing that revealed a homozygous NR2E3 c.119-2A>C variant, which disrupts the nearby universal “AG” splice acceptor site and is likely pathogenic.
S-cone ERG, acquired in response to a blue wavelength on an orange wavelength background, demonstrated a greater than anticipated response OU (Figure 6).
Epidemiology: Enhanced S-cone syndrome (ESCS) is a rare inherited progressive retinal degeneration. The prevalence of ESCS is not well-established; however, studies have found gene mutations associated with ESCS worldwide.1-12
Genetics: ESCS is inherited in an autosomal recessive pattern. NR2E3 (Nuclear Receptor Subfamily 2, Group E, Member 3) mutations are primarily implicated in ESCS. There are over 30 pathogenic mutations in NR2E3 that have been associated with ESCS.13-15 In the United States, however, the NR2E3 c.119-2A>C variant is the most common.16
Some ESCS cases have also documented NRL (neural retina leucine zipper) mutations.17-20
Pathophysiology: NR2E3 protein is a retinal orphan nuclear receptor, a transcription factor expressed in the outer nuclear layer of the human retina.13 To gain mechanistic insights, NR2E3 mutations in mouse models have been shown to disrupt the development of rod photoreceptors and disrupt the differentiation of cone photoreceptors to L/M-cones leading to over-expansion of the S-cone population in the retina.21-23 NR2E3 expression is under the direct regulation of the transcription factor, NRL protein.24 Therefore, mutations in NRL lead to ESCS by affecting NR2E3 expression.25
Clinical presentation: The clinical presentation of ESCS is variable; however, listed below are some documented symptoms.26-28
- Night blindness (nyctalopia, due to reduced functioning rod photoreceptors)
- Increased sensitivity to blue light (due to the over-expansion of functioning S-cones)
- Reduced visual acuity
- Abnormal color vision, with a tendency to see colors as more vibrant or intense
Diagnosis: Given that the clinical presentation of ESCS is variable, the diagnostic testing might also demonstrate variable findings among patients. Below are some diagnostic findings associated with each clinical test:
- Fundus exam – Fundus exam might demonstrate torpedo-like lesions primarily located along the vascular arcades, subretinal fibrosis, nummular pigmentary changes along the vascular arcades, or intraretinal yellow dots.6,29
- ERG – full-field ERG might demonstrate a diminished rod response and similar waveform in response to both scotopic and photopic conditions. The 30 Hz flicker responses might demonstrate significantly delayed responses with attenuated amplitudes. With orange background, increased response to short wavelength (blue) might be elicited. The multifocal ERG (mfERG) might show preserved central responses, though can be delayed.6,30-35
- OCT – OCT might demonstrate cystoid macular edema (CME) or foveomacular schisis.31 There are some documented cases of subretinal fibrosis secondary to choroidal neovascularization that might also be detected by OCT.6,35-27 There was also a case of macular retinal vascularization of the posterior pole that was detected by OCT.38
- FA – In the peripheral retina, FA might demonstrate hypo-autofluorescence with patchy areas of advanced hypo-autofluorescence. In the macula, hyper-autofluorescent flecks have been documented.6 FA might also identify vascular changes (i.e., retinal or choroidal neovascularization) and complement OCT findings.
- Genetic testing can identify mutations in the NR2E3 or NRL genes and confirm the diagnosis
Differential diagnosis: Nyctalopia has a broad differential and includes conditions such as Vitamin A deficiency, cataracts, autoimmune retinopathy, glaucoma, and other inherited retinal dystrophies (i.e., congenital stationary night blindness, Oguchi’s disease, fundus albipunctatus). Therefore, a careful workup is required to narrow the differential to treat patients appropriately.
Some NR2E3 mutations are implicated in autosomal dominant retinitis pigmentosa, Goldmann-Favre syndrome, and clumped pigmentary retinal degeneration.39-41 Characterizing the underlying disease caused by the NR2E3 mutations might not improve medical management but would inform genetic counseling.
Management: There are currently no approved therapies to treat ESCS. However, CRISPR/Cas9 gene editing has been used preclinically to correct the homozygous NR2E3 c.119-2A>C splice site variant in induced pluripotent stem cells (iPSCs) from two patients with ESCS.42 This finding suggests that CRISPR/Cas9 gene editing might be a plausible treatment approach for ESCS in the future.
Current medical management includes treating foveomacular schisis and CME with carbonic anhydrase inhibitors, while choroidal neovascularization is treated with agents that inhibit vascular endothelial growth factor.43-49 Genetic testing and patient support programs should also be offered in all patient cases. Genetic penetrance and carrier frequency of the NR2E3 mutations are not well understood given the rarity of disease phenotypes. Therefore, genetic testing of the patient and family might help provide further information to improve genetic counseling.
Summary of the Case:
- ESCS is a rare inherited progressive retinal degenerative disease of the photoreceptors that can present as nyctalopia
- There is clinical and diagnostic variability among patients with ESCS, but mutations in the transcription factors, NR2E3 or NRL, are implicated in pathology
- Current treatment is limited to treating foveomacular schisis and CME with carbonic anhydrase inhibitors, and choroidal neovascularization with anti-VEGF agents
- Genetic testing should be offered to inform genetic counseling
- Studies exploring CRISPR/Cas9 gene editing show promise for future therapies
References
- Bandah, D., Merin, S., Ashhab, M., Banin, E. & Sharon, D. The spectrum of retinal diseases caused by NR2E3 mutations in Israeli and Palestinian patients. Arch Ophthalmol 127, 297-302, doi:10.1001/archophthalmol.2008.615 (2009).
- Gao, F. J. et al. Genetic and Clinical Findings in a Large Cohort of Chinese Patients with Suspected Retinitis Pigmentosa. Ophthalmology 126, 1549-1556, doi:10.1016/j.ophtha.2019.04.038 (2019).
- Alsalamah, A. K., Khan, A. O., Bakar, A. A., Schatz, P. & Nowilaty, S. R. Recognizable Patterns of Submacular Fibrosis in Enhanced S-Cone Syndrome. Ophthalmol Retina 5, 918-927, doi:10.1016/j.oret.2021.03.014 (2021).
- Habibi, I. et al. Different Phenotypes in Pseudodominant Inherited Retinal Dystrophies. Front Cell Dev Biol 9, 625560, doi:10.3389/fcell.2021.625560 (2021).
- Al-Khuzaei, S. et al. Novel Pathogenic Sequence Variants in NR2E3 and Clinical Findings in Three Patients. Genes (Basel) 11, doi:10.3390/genes11111288 (2020).
- de Carvalho, E. R. et al. Enhanced S-Cone Syndrome: Spectrum of Clinical, Imaging, Electrophysiologic, and Genetic Findings in a Retrospective Case Series of 56 Patients. Ophthalmol Retina 5, 195-214, doi:10.1016/j.oret.2020.07.008 (2021).
- Hebbar, P. et al. Genetic risk variants for metabolic traits in Arab populations. Sci Rep 7, 40988, doi:10.1038/srep40988 (2017).
- Van Cauwenbergh, C. et al. Mutations in Splicing Factor Genes Are a Major Cause of Autosomal Dominant Retinitis Pigmentosa in Belgian Families. PLoS One 12, e0170038, doi:10.1371/journal.pone.0170038 (2017).
- Kuniyoshi, K. et al. New truncation mutation of the NR2E3 gene in a Japanese patient with enhanced S-cone syndrome. Jpn J Ophthalmol 60, 476-485, doi:10.1007/s10384-016-0470-0 (2016).
- Kannabiran, C., Singh, H., Sahini, N., Jalali, S. & Mohan, G. Mutations in TULP1, NR2E3, and MFRP genes in Indian families with autosomal recessive retinitis pigmentosa. Mol Vis 18, 1165-1174 (2012).
- Udar, N., Small, K., Chalukya, M., Silva-Garcia, R. & Marmor, M. Developmental or degenerative–NR2E3 gene mutations in two patients with enhanced S cone syndrome. Mol Vis 17, 519-525 (2011).
- Naik, A. et al. Enhanced S-cone syndrome: Clinical spectrum in Indian population. Indian J Ophthalmol 67, 523-529, doi:10.4103/ijo.IJO_1480_18 (2019).
- Haider, N. B. et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat Genet 24, 127-131, doi:10.1038/72777 (2000).
- Schorderet, D. F. & Escher, P. NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), Goldmann-Favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP). Hum Mutat 30, 1475-1485, doi:10.1002/humu.21096 (2009).
- Escher, P. et al. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum Mutat 30, 342-351, doi:10.1002/humu.20858 (2009).
- Stone, E. M. et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 124, 1314-1331, doi:10.1016/j.ophtha.2017.04.008 (2017).
- Iarossi, G. et al. A Novel Autosomal Recessive Variant of the NRL Gene Causing Enhanced S-Cone Syndrome: A Morpho-Functional Analysis of Two Unrelated Pediatric Patients. Diagnostics (Basel) 12, doi:10.3390/diagnostics12092183 (2022).
- Newman, H. et al. Homozygosity for a Recessive Loss-of-Function Mutation of the NRL Gene Is Associated With a Variant of Enhanced S-Cone Syndrome. Invest Ophthalmol Vis Sci 57, 5361-5371, doi:10.1167/iovs.16-19505 (2016).
- Nishiguchi, K. M. et al. Recessive NRL mutations in patients with clumped pigmentary retinal degeneration and relative preservation of blue cone function. Proc Natl Acad Sci U S A 101, 17819-17824, doi:10.1073/pnas.0408183101 (2004).
- Littink, K. W. et al. Autosomal Recessive NRL Mutations in Patients with Enhanced S-Cone Syndrome. Genes (Basel) 9, doi:10.3390/genes9020068 (2018).
- Haider, N. B., Naggert, J. K. & Nishina, P. M. Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum Mol Genet 10, 1619-1626, doi:10.1093/hmg/10.16.1619 (2001).
- Haider, N. B. et al. Nr2e3-directed transcriptional regulation of genes involved in photoreceptor development and cell-type specific phototransduction. Exp Eye Res 89, 365-372, doi:10.1016/j.exer.2009.04.006 (2009).
- Haider, N. B. et al. The transcription factor Nr2e3 functions in retinal progenitors to suppress cone cell generation. Vis Neurosci 23, 917-929, doi:10.1017/S095252380623027X (2006).
- Oh, E. C. et al. Rod differentiation factor NRL activates the expression of nuclear receptor NR2E3 to suppress the development of cone photoreceptors. Brain Res 1236, 16-29, doi:10.1016/j.brainres.2008.01.028 (2008).
- Mears, A. J. et al. Nrl is required for rod photoreceptor development. Nat Genet 29, 447-452, doi:10.1038/ng774 (2001).
- Khan, A. O., Aldahmesh, M. & Meyer, B. The enhanced S-cone syndrome in children. BMJ Case Rep 2009, doi:10.1136/bcr.10.2008.1163 (2009).
- Fishman, G. A., Jampol, L. M. & Goldberg, M. F. Diagnostic features of the Favre-Goldmann syndrome. Br J Ophthalmol 60, 345-353, doi:10.1136/bjo.60.5.345 (1976).
- Garafalo, A. V. et al. Cone Vision Changes in the Enhanced S-Cone Syndrome Caused by NR2E3 Gene Mutations. Invest Ophthalmol Vis Sci 59, 3209-3219, doi:10.1167/iovs.18-24518 (2018).
- Yzer, S. et al. Expanded clinical spectrum of enhanced S-cone syndrome. JAMA Ophthalmol 131, 1324-1330, doi:10.1001/jamaophthalmol.2013.4349 (2013).
- Tsang, S. H. & Sharma, T. Enhanced S-Cone Syndrome (Goldmann-Favre Syndrome). Adv Exp Med Biol 1085, 153-156, doi:10.1007/978-3-319-95046-4_28 (2018).
- Vincent, A., Robson, A. G. & Holder, G. E. Pathognomonic (diagnostic) ERGs. A review and update. Retina 33, 5-12, doi:10.1097/IAE.0b013e31827e2306 (2013).
- Hood, D. C., Cideciyan, A. V., Roman, A. J. & Jacobson, S. G. Enhanced S cone syndrome: evidence for an abnormally large number of S cones. Vision Res 35, 1473-1481, doi:10.1016/0042-6989(95)98727-q (1995).
- Jacobson, S. G., Marmor, M. F., Kemp, C. M. & Knighton, R. W. SWS (blue) cone hypersensitivity in a newly identified retinal degeneration. Invest Ophthalmol Vis Sci 31, 827-838 (1990).
- Marmor, M. F., Jacobson, S. G., Foerster, M. H., Kellner, U. & Weleber, R. G. Diagnostic clinical findings of a new syndrome with night blindness, maculopathy, and enhanced S cone sensitivity. Am J Ophthalmol 110, 124-134, doi:10.1016/s0002-9394(14)76980-6 (1990).
- Audo, I. et al. Phenotypic variation in enhanced S-cone syndrome. Invest Ophthalmol Vis Sci 49, 2082-2093, doi:10.1167/iovs.05-1629 (2008).
- Nakamura, M. et al. Enhanced S-cone syndrome with subfoveal neovascularization. Am J Ophthalmol 133, 575-577, doi:10.1016/s0002-9394(01)01428-3 (2002).
- Zerbib, J. et al. Retinochoroidal Anastomosis Associated with Enhanced S-Cone Syndrome. Retin Cases Brief Rep 13, 295-299, doi:10.1097/ICB.0000000000000594 (2019).
- Bazvand, F., Khojasteh, H. & Zarei, M. Novel findings in enhanced S-cone syndrome: a case with macular retinal neovascularization and severe retinal vasculitis. Doc Ophthalmol 139, 221-226, doi:10.1007/s10633-019-09706-6 (2019).
- Sharon, D., Sandberg, M. A., Caruso, R. C., Berson, E. L. & Dryja, T. P. Shared mutations in NR2E3 in enhanced S-cone syndrome, Goldmann-Favre syndrome, and many cases of clumped pigmentary retinal degeneration. Arch Ophthalmol 121, 1316-1323, doi:10.1001/archopht.121.9.1316 (2003).
- Chavala, S. H. et al. An Arg311Gln NR2E3 mutation in a family with classic Goldmann-Favre syndrome. Br J Ophthalmol 89, 1065-1066, doi:10.1136/bjo.2005.068130 (2005).
- Gire, A. I. et al. The Gly56Arg mutation in NR2E3 accounts for 1-2% of autosomal dominant retinitis pigmentosa. Mol Vis 13, 1970-1975 (2007).
- Bohrer, L. R. et al. Correction of NR2E3 Associated Enhanced S-cone Syndrome Patient-specific iPSCs using CRISPR-Cas9. Genes (Basel) 10, doi:10.3390/genes10040278 (2019).
- Iannaccone, A., Fung, K. H., Eyestone, M. E. & Stone, E. M. Treatment of adult-onset acute macular retinoschisis in enhanced s-cone syndrome with oral acetazolamide. Am J Ophthalmol 147, 307-312 e302, doi:10.1016/j.ajo.2008.08.003 (2009).
- Chatzistergiou, V. et al. Optical Coherence Tomography Analysis of Cystoid Macular Edema in Retinal Dystrophy Treated with Oral Acetazolamide: Two Cases. Klin Monbl Augenheilkd 237, 484-486, doi:10.1055/a-1068-2762 (2020).
- Genead, M. A., Fishman, G. A. & McAnany, J. J. Efficacy of topical dorzolamide for treatment of cystic macular lesions in a patient with enhanced S-cone syndrome. Doc Ophthalmol 121, 231-240, doi:10.1007/s10633-010-9247-9 (2010).
- Hajali, M. & Fishman, G. A. Dorzolamide use in the management of macular cysts in a patient with enhanced s-cone syndrome. Retin Cases Brief Rep 3, 121-124, doi:10.1097/ICB.0b013e31818faa21 (2009).
- Bechet, L. et al. Management of a case of Enhanced S-cone syndrome with massive foveoschisis treated with pars plana vitrectomy with silicone oil tamponade. Ophthalmic Genet 42, 615-618, doi:10.1080/13816810.2021.1925927 (2021).
- Broadhead, G. K., Grigg, J. R., McCluskey, P., Korsakova, M. & Chang, A. A. Bevacizumab for choroidal neovascularisation in enhanced S-cone syndrome. Doc Ophthalmol 133, 139-143, doi:10.1007/s10633-016-9555-9 (2016).
- Bertoli, F., Pignatto, S., Rizzetto, F. & Lanzetta, P. A 5-Year-Old Case of Choroidal Neovascularization in Enhanced S-Cone Syndrome Treated with Ranibizumab. Case Rep Ophthalmol 9, 510-515, doi:10.1159/000495743 (2018).
Faculty Approval by: Paul Bernstein, MD, PhD
Copyright: Copyright Ramshekar and Ronquillo, ©2023. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Identifier: Moran_CORE_126913
Lyme Disease-Associated Uveitis: A Case Report and Review Emphasizing the Importance of Travel History and Geographic Considerations
Home / Intraocular Inflammation and Uveitis / Infectious Uveitis
Title: Lyme Disease-Associated Uveitis: A Case Report and Review Emphasizing the Importance of Travel History and Geographic Considerations
Authors: Andrew DesLauriers, BA; Marissa Larochelle, MD
Photographer: Rich Ordonez
Date: 08/09/2023
Keywords/Main Subjects: lyme disease, intermediate uveitis, anterior uveitis, travel history
Diagnosis: Lyme Disease-Associated Intermediate and Anterior Uveitis
Description of Case:
Introduction:
Lyme disease, caused by the Borrelia genus of bacteria transmitted through Ixodes ticks, is primarily reported in the Northeast and Mid-Atlantic regions of the United States, as well as in certain parts of Europe and Japan.1,2 Ocular involvement can occur at any stage of the disease, presenting with diverse manifestations, including uveitis. Early identification of Lyme-associated uveitis is crucial to initiate appropriate treatment and prevent long-term complications. Lyme-associated uveitis should be considered in the differential diagnosis of patients with new-onset uveitis and a history of travel to endemic areas.
Case Presentation:
In early August, a 10-year-old male presented to the Moran Eye Center in Utah with sharp, bilateral eye pain, blurred vision, and increased redness for three days. He reported a history of mild, intermittent eye redness without pain for three months prior to presentation. Additionally, one month before the increase in ocular symptoms, the patient experienced a brief generalized illness with fevers, headache, and cervical/post-cervical and submandibular lymphadenopathy, initially attributed to a self-limited viral illness. The patient, who had recently been diagnosed with autoimmune thyroiditis and a strong family history of autoimmune diseases, reported no previous ocular history. The patient’s mother reported that he regularly interacts with several stray cats near their home, but there were no instances of reported bites or scratches. Additionally, the patient and his family had vacationed in Vermont, a Lyme disease endemic area, three months prior to his presentation at the Moran Eye Center, though no tick bites were reported during their trip.
Examination and Diagnosis:
On examination, the patient had 1-2+ mixed cells in the anterior chamber and 1+ cell and 2+ vitreous haze bilaterally with notable vitreous debris. Fluorescein angiography revealed mild diffuse vascular leakage in both eyes. Based on the clinical picture, the patient was diagnosed with bilateral, simultaneous anterior and intermediate uveitis. Initial laboratory workup was significant for positive Bartonella IgM (1:256) but negative Bartonella IgG. Consequently, the patient was treated for presumed Bartonella infection with doxycycline and rifampin, along with topical prednisolone, cyclopentolate, and an oral prednisone taper to manage inflammation and prevent complications. The patient was referred to Infectious Disease clinic and further history taking revealed hiking in Lyme-endemic Vermont, so Lyme serology was added to the lab work up.
Treatment and Follow-Up:
Two weeks later, additional lab results indicated a positive Lyme ELISA and Immunoblot assay. While the initial Bartonella IgM had been elevated, a repeat Bartonella IgM test was negative (1:64), and two Bartonella IgG assays were also both negative, indicating that the initial positive IgM was likely a false-positive. Fortunately, doxycycline is effective in the treatment of both Bartonella and Lyme Borreliosis, so the patient had already been started on an effective regimen. Rifampin was discontinued, and the patient continued doxycycline for an additional 6 weeks. Following a two-month course of doxycycline with an oral prednisone taper, the patient’s symptoms had completely resolved, and a clinical examination showed no remaining signs of intraocular inflammation. The patient remained symptom-free, with no intraocular inflammation or recurrences at 11 months after the initial presentation.
Lyme Disease Overview:
The Borrelia genus of bacteria, transmitted through a bite from an Ixodes tick, cause Lyme disease, which progresses through three clinical stages. Stage 1 is characterized by a bulls-eye rash (Erythema Migrans) at the infection site in around 80% of patients, accompanied by constitutional flu-like symptoms in some cases. Stage 2 involves hematogenous spread to organs, leading to a variety of sequalae, including meningitis, neuropathies, and acute carditis. Stage 3 presents months to years post-inoculation with chronic manifestations, most commonly oligoarticular arthritis. Notably, 2-3% of Lyme disease patients present with Stage 2 or Stage 3 sequelae within days of exposure.3 Infection with Borrelia tends to peak during the late spring or early summer in endemic areas.3
Ocular Involvement in Lyme Disease:
Ocular involvement, typically bilateral, can occur at any disease stage.4 The most common ocular symptom associated with Lyme disease is a follicular conjunctivitis, occurring in approximately 11% of patients most commonly during Stage 1 of disease.5 Stages 2 and 3 of Lyme disease are associated with a variety of symptoms including all types of uveitis, retinal vasculitis, neuroretinis, episcleritis, keratitis, papillitis, optic neuritis, and cranial nerve palsies of nerves III, V, VI, and VII.4,6
Lyme-Associated Uveitis:
Lyme uveitis, an uncommon manifestation of Stage 2 and Stage 3 Lyme disease, comprises up to 4.3% of all uveitis cases at referral centers in endemic areas; however, the reported prevalence varies widely and is much lower in non-endemic areas.4,7 Patients typically present with symptoms such as eye pain, redness, blurred vision, new floaters, and photophobia. The most common presentation is an intermediate uveitis; however, cases of anterior, posterior, and panuveitis have also been reported.4 Uveitis is commonly accompanied by retinal vasculitis. The uveitis can be granulomatous or non-granulomatous in nature.8 Published reports have described varied clinical findings of Lyme uveitis in different patients, with some demonstrating choroidal neovascularization and others showing multifocal white dots in the posterior pole, resembling white dot syndromes such as acute posterior multifocal placoid pigment epitheliopathy (APMPPE).9,10,11
Diagnosis and Management:
Lyme-associated uveitis should be considered in any patient presenting with new-onset uveitis after travel to a Lyme-endemic area. A detailed history should focus on possible exposure and any symptoms outside of eye-related issues, though ocular involvement can be the sole manifestation of Lyme infection, meaning patients may not have other systemic symptoms.4,6 Particular attention should be paid to patients who report spending any amount of time outdoors in an endemic region, engaging in activities such as hiking, gardening, hunting, or forestry work.3
Tissue biopsies are seldom useful in the detection of ocular Borrelia infection.4 Routine screening for Lyme disease in all uveitis patients isn’t recommended due to the low incidence of Lyme-associated uveitis. However, in the right clinical context and with a history of travel to Lyme-endemic areas, serum antibody testing can be used to confirm the diagnosis. Lyme-associated uveitis management should include regular surveillance, and multi-modal imaging is useful for monitoring progression and treatment response.11
Treatment:
Borrelis burgdorferi, the species of bacteria causing Lyme disease in the United States, is typically sensitive to tetracyclines and many B-lactam antibiotics. Doxycycline, ceftriaxone, and amoxicillin can treat any stage of disease, with ceftriaxone preferred for CNS involvement.3,4 These medications are also effective in treating ocular disease, with some studies suggesting use of a cephalosporin for better ocular penetration.12 Steroid treatment alone does not worsen disease but is insufficient in resolving ocular complications.4 Lyme-associated uveitis typically requires a combined approach of both antibiotic treatment and management of inflammation.
Discussion:
This case illustrates the importance of maintaining a high suspicion for Lyme disease in patients with uveitis if there is a report of recent travel to Lyme-endemic areas. Conversely, it is important to note that Lyme disease is a less likely cause of uveitis in patients without exposure to an endemic region, for example, residents of Utah who have not travelled out of the state. While there are small populations of ticks in Utah that could potentially carry Lyme disease, tests conducted on samples of these ticks have not detected the presence of Borrelia bacteria.13 Even in endemic areas, Lyme disease is an uncommon cause of uveitis, however it should be considered in patients with the appropriate risk factors such as a history of time spent outdoors and additional clinical symptoms. Please see the geographic distributions of reported Lyme disease included in the resources section below to help guide risk stratification based on travel history.
Lyme-associated uveitis can present in various forms, making it crucial to include this infectious etiology in the differential diagnosis when risk factors are present and perform appropriate antibody testing for confirmation. In this case, the patient’s travel to an endemic area in late spring placed him at risk of Lyme exposure. His intermittent eye redness preceding the acute uveitis episode was likely attributable to the follicular conjunctivitis often reported in Lyme disease’s Stage 1. Furthermore, the febrile illness he experienced one month prior to presenting at the Moran Eye Center was likely a systemic manifestation of Lyme disease. Early identification allows for timely treatment and prevents vision-threatening complications.14 Untreated Lyme uveitis, although rare, can result in complete loss of vision and phthisis bulbi.15
Conclusion:
Lyme-associated uveitis, though uncommon, can lead to serious complications. This case report emphasizes the significance of clinical acumen in diagnosing and treating vision-threatening diseases related to Lyme borreliosis. Awareness of Lyme disease’s geographic distribution and risk factors can guide risk stratification based on travel history, helping to identify cases promptly and initiate appropriate management. Clinicians in non-endemic areas should maintain a high index of suspicion for Lyme uveitis in patients with relevant travel history, particularly in the setting of characteristic systemic symptoms.
Images or video:

Figure 1: Lyme Disease Map. Distribution of reported lyme disease cases in the United States in 2021. Each green dot represents a case of Lyme Disease reported to the CDC in 2021. States shaded with light blue are regarded as “high incidence states” by the CDC. States shaded in grey are considered low incidence. Lyme Disease Map acquired from the CDC website: https://www.cdc.gov/lyme/datasurveillance/lyme-disease-maps.html


Figure 2. Fluorescein angiography images of the patient’s right eye that were taken upon presentation. The images captured at 0:35 (top) and 5:24 (bottom) after infusion reveal mild diffuse vascular leakage.
Summary of the Case:
Lyme disease, caused by the Borrelia genus of bacteria and transmitted by Ixodes ticks, can lead to a variety of ocular complications including uveitis. Transmission typically occurs in endemic regions with the appropriate ecological factors to allow for bacterial reproduction and transmission. A 10-year-old male presented with eye pain, redness, and blurred vision after vacationing in a Lyme-endemic region, although he had no reported tick bites. His examination and subsequent tests revealed Lyme-associated uveitis. The importance of early identification and treatment is underscored, as untreated cases can result in severe vision complications. Clinicians in non-endemic regions should maintain vigilance for Lyme uveitis in patients with relevant travel history in order to ensure prompt and effective management.
Format: Case Report, Literature Review
References
- Lyme Disease Map | Lyme Disease | CDC. Accessed August 1, 2023. https://www.cdc.gov/lyme/datasurveillance/lyme-disease-maps.html
- Overview | Johns Hopkins Lyme and Tickborne Diseases Dashboard. Accessed August 1, 2023. https://www.hopkinslymetracker.org/overview/
- Steere AC, Strle F, Wormser GP, et al. Lyme borreliosis. Nature Reviews Disease Primers. 2016;2(1):16090. doi:10.1038/nrdp.2016.90
- Bernard A, Seve P, Abukhashabh A, et al. Lyme-associated uveitis: Clinical spectrum and review of literature. Eur J Ophthalmol. 2020;30(5):874-885. doi:10.1177/1120672119856943
- Steere AC, Bartenhagen NH, Craft JE, et al. The early clinical manifestations of Lyme disease. Ann Intern Med. 1983;99(1):76-82. doi:10.7326/0003-4819-99-1-76
- Lesser RL. Ocular manifestations of Lyme disease. The American Journal of Medicine. 1995;98(4, Supplement 1):60S-62S. doi:10.1016/S0002-9343(99)80045-X
- Mikkilä H, Seppälä I, Leirisalo-Repo M, Immonen I, Karma A. The etiology of uveitis: The role of infections with special reference to Lyme borreliosis. Acta Ophthalmologica Scandinavica. 1997;75(6):716-719. doi:10.1111/j.1600-0420.1997.tb00637.x
- Mikkilä HO, Seppälä IJT, Viljanen MK, Peltomaa MP, Karma A. The expanding clinical spectrum of ocular lyme borreliosis. Ophthalmology. 2000;107(3):581-587. doi:10.1016/S0161-6420(99)00128-1
- Kılıç Müftüoğlu İ, Aydın Akova Y, Gür Güngör S. A Case of Lyme Disease Accompanied by Uveitis and White Dot Syndrome. Turk J Ophthalmol. 2016;46(5):241-243. doi:10.4274/tjo.25991
- Amer R, Brannan S, Forrester JV. Inflammatory choroidal neovascular membrane in presumed ocular Lyme borreliosis. Acta Ophthalmol. 2009;87(3):346-348. doi:10.1111/j.1755-3768.2007.01160.x
- Ferro Desideri L, Rosa R, Forte P, et al. Multimodal imaging for the management of Lyme-associated uveitis: A case report from an Italian tertiary center. European Journal of Ophthalmology. Published online January 29, 2023:11206721231154172. doi:10.1177/11206721231154172
- Lindström BE, Skogman BH, Lindström AK, Tallstedt L, Nilsson K. Borrelia Ocular Infection: A Case Report and a Systematic Review of Published Cases. Ophthalmic Res. 2022;65(2):121-130. doi:10.1159/000521307
- Richardson K, Davis R, Ramirez R. Ticks and Tick-Borne Diseases of Utah. Utah State University Extension and Utah Plant Pest Diagnostic Laboratory; 2023.
- Copeland RAJ. Lyme uveitis. Int Ophthalmol Clin. 1990;30(4):291-293. doi:10.1097/00004397-199030040-00019
- Kauffmann DJ, Wormser GP. Ocular Lyme disease: case report and review of the literature. British Journal of Ophthalmology. 1990;74(6):325-327. doi:10.1136/bjo.74.6.325
Faculty Approval by: Marissa Larochelle, MD
Copyright: Andrew DesLauriers, ©2023. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Identifier: Moran_CORE_126899
Iridocorneal Endothelial (ICE) Syndrome
Title: Iridocorneal Endothelial (ICE) Syndrome
Author: Krishna Mallem, Drexel University College of Medicine, MD Class of 2024; . Austin Nakatsuka, MD
Date: July 2023
Keywords/Main Subjects: Iridocorneal endothelial syndrome; ICE; Gonioscopy assisted transluminal trab
-

- Electron microscopy of corneal endothelium in ICE syndrome.

-

- Polycoria, corectopia and atrophic iris changes in a patient.

eculotomy; GATT
Introduction:
Iridocorneal endothelial (ICE) syndrome is an unusual disorder of the eye that involves an abnormality of the corneal endothelium. ICE syndrome typically affects middle aged women, and occurs sporadically and unilaterally 1,2. It consists of three clinical variants, which are Chandler syndrome, essential iris atrophy, and iris nevus or Cogan-Reese Syndrome. The distinction between these variants is based on clinical exam findings. Abnormalities of the corneal endothelium in ICE syndrome can lead to varying degrees of corneal edema, iris atrophy, and secondary angle closure glaucoma. This condition can be potentially blinding due to glaucoma recalcitrant to conventional therapies and recurrent corneal edema1-4. We present a case of a patient with ICE syndrome who undergoes gonioscopy assisted transluminal trabeculotomy (GATT) for uncontrolled IOP on medical therapy.
Photos taken from Moran Eye Center archives.
Pathophysiology:
Similar to corneal guttata found in Fuchs endothelial dystrophy, the corneal endothelium in patients with ICE syndrome is described to have a “beaten bronze” appearance. Scanning and tunneling electron microscopy of endothelial cells in patients with ICE syndrome reveal a population of well differentiated epithelial-like cells in place of normal endothelium. This cell population displays migratory characteristics, and is believed to migrate posteriorly onto the trabecular meshwork and the iris. As this tissue contracts within the angle and the iris, peripheral anterior synechiae (PAS) may form, and the iris may undergo morphological changes. Contraction of tissue can also predispose patients to secondary angle closure glaucoma. Corneal edema can develop as a consequence of elevated intraocular pressure as well as due to failure of the normal pump function of the dysfunctional endothelium3-8.
Case presentation:
Our patient is a 51 year old male who presented in February 2017 for glaucoma evaluation in the left eye (OS). IOP on examination was 34. Corneal pachymetry showed a corneal thickness of 523 microns OS. Gonioscopy revealed a broad area of peripheral anterior synechiae (PAS) temporally, with smaller areas of PAS inferiorly. Slit lamp exam was significant for diffuse endothelial guttae-like changes in the cornea with an oval shaped pupil. IOP and slit lamp exam of the right eye were unremarkable. Baseline visual fields were obtained and showed scattered defects in both eyes. Baseline optical coherence tomography (OCT) of the retinal nerve fiber layer (RNFL) revealed normal thickness in both eyes. Given examination findings at the baseline visit, the patient was diagnosed with ICE syndrome and was started on latanoprost once daily OS for intraocular pressure control.
The patient was monitored regularly and over the course of several years his medical therapy was escalated due to persistently high IOPs as well as changes on field and OCT testing. In March 2022, the patient was on maximal medical therapy with netarsudil-latanoprost, dorzolamide-timolol, and brimonidine in the left eye. Despite this, his IOP was 37 on the left. Slit lamp examination at this time showed a distorted pupil with high PAS temporally. Visual field testing showed a subtle superior arcuate scotoma OS. OCT RNFL showed significant thinning inferiorly and superiorly OS. The decision to proceed with surgery was made given the patient’s young age and long term prognosis due to medically uncontrolled IOP.
Patient underwent gonioscopy assisted transluminal trabeculotomy (GATT) with anterior goniosynechiolysis without cataract extraction in May of 2022. After successful surgery without complications, the patient’s iop quickly normalized and was maintained over a year later on two drops. OCT findings 6 months after surgery revealed stability, however, there were signs of progression 1 year following the procedure, despite well controlled IOPs. Additionally, the patient has recently developed corneal guttae and edema which is currently being treated medically with hypertonic . Although the patient’s IOP continues to be well controlled, the current focus is now on controlling corneal edema, with consideration for corneal transplantation in the future if the cornea does not recover with medical therapy alone.
Discussion:
This is a unique case of a patient with ICE syndrome who was managed with GATT and goniosynechiolysis in the left eye for uncontrolled IOP and progressive glaucomatous changes. To our knowledge, there is not another case of GATT or other microinvasive glaucoma surgery (MIGS) being performed in a patient with ICE syndrome in the literature. It is well documented that filtering procedures in ICE syndrome are only modestly successful at 5 years (53% with drainage device and 29% with trabeculectomy)⁸ and often need to be repeated to obtain IOP control. Although GATT has successfully controlled IOP one year out from surgery, there is still evidence of OCT progression and further surgical treatment may be necessary. Furthermore, the patient has developed more clinically significant corneal edema and guttae, suggesting corneal decompensation, which may require surgical intervention with a corneal transplant. This case highlights the difficulty in managing patients with ICE syndrome and documents the novel use of GATT for IOP control in ICE syndrome.
References:
- Laganowski, H. C., Muir, M. G. K., & Hitchings, R. A. (1992). Glaucoma and the iridocorneal endothelial syndrome. Archives of Ophthalmology, 110(3), 346-350.
- Dubey, S., Jain, K., Singh, S., & Mukhejee, S. (2021). Iridocorneal Endothelial Syndrome with Coexisting Macular Edema and Neurosensory Detachment: An Unusual Case Report. Journal of Current Glaucoma Practice, 15(3), 149.
- Campbell, D. G., Shields, M. B., & Smith, T. R. (1978). The corneal endothelium and the spectrum of essential iris atrophy. American journal of ophthalmology, 86(3), 317-324.
- Basic and Clinical Science Course (BCSC). American Academy of Ophthalmology, (2010-2011), 142-144.
- Eagle, R. C., Font, R. L., Yanoff, M., & Fine, B. S. (1979). Proliferative endotheliopathy with iris abnormalities: the iridocorneal endothelial syndrome. Archives of Ophthalmology, 97(11), 2104-2111.
- Doan A, Alward W: Iridocorneal Endothelial Syndrome (ICE) – essential iris atrophy : 63-year-old female with PAS, “iris mass”, corectopia, and increased IOP OS. February 21, 2005; Available from: http://www.EyeRounds.org/cases/case14.htm.
- Levy, S. G., Kirkness, C. M., Moss, J., Ficker, L., & McCartney, A. C. (1996). The histopathology of the iridocorneal-endothelial syndrome. Cornea, 15(1), 46-54.
- Doe, E. A., Budenz, D. L., Gedde, S. J., & Imami, N. R. (2001). Long-term surgical outcomes of patients with glaucoma secondary to the iridocorneal endothelial syndrome. Ophthalmology, 108(10), 1789-1795.
Intraocular Lens (IOL) Opacification and Calcification in Patients with Hydrophilic Lenses
Title: Intraocular Lens (IOL) Opacification and Calcification in Patients with Hydrophilic Lenses
Author (s): Cuneyt Ozkardes, MS; Kevin Eid, MD; Austin Nakatsuka, MD
Date: 7/20/2023
Keywords/Main Subjects: IOL; cataract complications; hydrophilic lens; calcification.
Diagnosis: Calcification of a hydrophilic intraocular lens.
Summary of the Case:
- Intraocular Lens (IOL) opacification is a late post-operative complication, often caused by calcification of the lens material, particularly in hydrophilic lenses.
- This presentation can easily be mistaken for posterior capsule opacification (PCO), therefore, it is important to conduct a thorough ocular history and exam, especially in eyes with hydrophilic IOLs.
- The only treatment option currently for this condition is lens exchange surgery.
Description of Case:
Introduction
Intraocular Lens (IOL) opacification is a late post-operative complication of cataract surgery, especially in patients with hydrophilic lenses.1 The most common cause of opacification is calcification of the lens material.2 Proposed causes of this phenomenon include reactions with post-operative intracameral substances (i.e. Silicone, gas, viscoelastic), metabolic changes in the anterior chamber, or inflammatory reactions caused by surgical procedures.3-5 The only known treatment for this phenomenon is explantation with or without exchanging of the lens.6,7 In addition to their association with lens calcification, meta-analyses have shown hydrophilic lenses tend to have higher PCO rates compared to lenses of other materials.8 Because of this known complication associated with hydrophilic lens material, the most common implanted lenses are currently made of hydrophobic material.
Case Presentation
An 83-year-old male patient with a known history of pseudoexfoliation glaucoma in both eyes presented to our clinic with complaints of slowly worsening blurry and limited vision in the right eye over the past six months. Despite these symptoms, previous slit lamp examinations during this period had been unremarkable with BCVA of 20/20 bilaterally. The patient had undergone trabeculectomy in the right eye 21 months prior, which was complicated by a choroidal hemorrhage and subsequent choroidal detachment one week later. He was treated conservatively with prednisolone and atropine drops daily, as well as oral prednisone. His visual acuity improved from 20/300 at presentation to 20/20 after 8 weeks of treatment.
Upon examination during the current visit, BCVA in the right eye was 20/20. Slit-lamp examination revealed a central cloudy opacity in the posterior chamber intraocular lens (PCIOL), with haptics positioned at 2 and 8 o’clock. The left eye showed no signs of opacification or other complications. Based on the clinical findings and the significant impact on visual function, the decision was made to proceed with an IOL exchange surgery to address the opacification using a hydrophobic TECNIS Eyhance IOL.
The original IOL, placed nearly 13 years prior, was removed and sent for pathological analysis. It was identified as an Akreos Bausch and Lomb one-piece [plate] hydrophilic acrylic posterior capsule IOL (Model MI60L). The pathology report described the specimen as having a deposition of a white granular material on the anterior surface of the IOL in the center of the optic. The deposition was consistent with calcium and was specifically deposited centrally, denoting the area where the anterior capsulotomy is open. The peripheral part of the optic as well as the footplates showed no deposition of this material.
Follow-up examination eleven weeks after the IOL exchange surgery revealed a well-centered IOL with mild posterior capsule opacification (PCO). The patient’s BCVA was 20/20, with the patient noting he was much happier with his vision and seeing well. He expressed overall satisfaction with the new lens and reported no further visual complaints. Continued monitoring and regular follow-up visits were scheduled to assess the long-term stability of the IOL and to manage any potential future complications.
Images or video:

Figure 1. Slit-lamp examination revealing central cloudy opacification of the intraocular lens (IOL ).

Figure 2. Extracted Akreos Bausch and Lomb hydrophilic acrylic posterior capsule IOL (Model MI60L).

Figure 3. Close-up image of extracted capsule demonstrating central opacification .

Figure 4. Enhanced view revealing widespread calcification (seen in the image as diffuse round globules ) coating the central and anterior surfaces of the extracted IOL.
Format: Case Report
References:
- Grzybowski, A., Markeviciute, A., & Zemaitiene, R. (2020). A narrative review of intraocular lens opacifications: Update 2020. Annals of Translational Medicine, 8(22).
- Drimtzias, E. G., Rokidi, S. G., Gartaganis, S. P., & Koutsoukos, P. G. (2011). Experimental investigation on mechanism of hydrophilic acrylic intraocular lens calcification. American Journal of Ophthalmology, 152, 824-833.
- Werner, L., Wilbanks, G., Nieuwendaal, C. P., et al. (2015). Localized opacification of hydrophilic acrylic intraocular lenses after procedures using intracameral injection of air or gas. Journal of Cataract and Refractive Surgery, 41, 199-207.
- Stanojcic, N., Hull, C., & O’Brart, D. P. (2020). Clinical and material degradations of intraocular lenses: A review. European Journal of Ophthalmology, 30, 823-839.
- Pérez-Vives, C. (2018). Biomaterial Influence on Intraocular Lens Performance: An Overview. Journal of Ophthalmology, 2018, 2687385.
- Haymore, J., Zaidman, G., Werner, L., et al. (2007). Misdiagnosis of hydrophilic acrylic intraocular lens optic opacification: Report of 8 cases with the MemoryLens. Ophthalmology, 114, 1689-1695.
- Werner, L. (2007). Causes of intraocular lens opacification or discoloration. Journal of Cataract and Refractive Surgery, 33, 713-726.
- Findl, O., Buehl, W., Bauer, P., & Sycha, T. (2010). Interventions for preventing posterior capsule opacification. Cochrane Database of Systematic Reviews, 2. CD003738.
Faculty Approval by: Austin Nakatsuka, MD
Identifier: Moran_CORE_126859
Copyright Cuneyt Ozkardes, ©2023. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Congenital Ectropion Uveae
Title: Congenital Ectropion Uveae
Author (s): Samareh Dadashazar, MS IV, Texas Tech University Health Sciences Center School of Medicine
Photographer: James Gilman, CRA, FOPS
Keywords/Main Subjects: Congenital Ectropion Uveae; Secondary Glaucoma
Diagnosis: Congenital Ectropion Uveae
Author: Samareh Dadashazar, MS IV, Texas Tech University Health Sciences Center School of Medicine


Ectropion uveae is a condition where there is migration of the posterior iris pigmented epithelium from the pupillary ruff onto the anterior surface of the iris. This process can either be congenital or acquired. The congenital form is noted to be rare and non-progressive.1 An exact mechanism is currently unknown, but many theories have postulated that a late developmental arrest of posterior neural crest cells plays a role.1,3 Others suggest that there may be a type of primordial endothelium in the anterior chamber that does not fully regress and subsequently induces a hyperplasia of the pigmented epithelium leading to the anterior movement.2,4 In contrast, the acquired form occurs when a membrane develops in the anterior segment secondary to any inflammatory, neoplastic, or ischemic process.1 This membrane eventually contracts, pulling the posterior pigmented epithelium of the iris anteriorly, thus forming the ectropion uvea. As such, the acquired type is considered progressive in nature. The most common causes of acquired ectropion uvea are neovascular glaucoma and neovascularization of the iris.2,3,4
Congenital ectropion uvea is usually a unilateral finding, but bilateral cases have been reported in the literature.4 The ectropion uvea can be an isolated finding, seen alongside ptosis with good levator function, or as part of a systemic disorder such as neurofibromatosis type 1 (NF1), Rieger’s anomaly, and Prader-Willi syndrome.1–5 An extremely important association to be aware of is the one between congenital ectropion uvea and secondary glaucoma. The literature reports up to 80-90% of patients with congenital ectropion uvea will eventually develop glaucoma at some point in their life.2,5 There are approximately 50 established cases of glaucoma secondary to congenital ectropion uvea in the literature.5 Most patients are affected in childhood or early adolescence, but the reported ages have ranged from 7 months to 42 years.2,4,5 The variation in age of presentation likely correlates to the degree of neural crest cell arrest and trabecular meshwork malformation.2,4 While there are a couple of exceptions reported in the literature, the trend seems to be that patients with both congenital ectropion uvea and NF1 develop an angle-closure glaucoma while those without NF1 develop an open-angle glaucoma.1,3,5 In most cases of congenital ectropion uvea without NF1, patients have anterior insertion of the iris, typically at the level of the trabecular meshwork but sometimes as anterior as Schwalbe’s line.2,5 With such anterior insertion, it is clear to see how these open but dysplastic angles predispose these patient to develop glaucoma over time.
Currently, there is not a consensus on what the best treatment is for patients with glaucoma secondary to congenital ectropion uvea. In contrast to primary congenital glaucoma, many articles in the literature have reported that goniotomies and trabeculotomies are not effective for long-term pressure control and that patients who received these interventions subsequently needed a trabeculectomy or cycloablation in order to bring their pressures back down.2,5 The longest reported case of intraocular pressure control due to goniotomy alone is only 2.5 years.5 Glaucoma drainage devices also seem to fail at controlling pressures in these patients.5 Currently, filtering procedures with antifibrotics, such as a trabeculectomy with mitomycin c, seem to be the most successful surgical intervention for managing long-term intraocular pressures in patients with congenital ectropion uvea.5
In conclusion, congenital ectropion uvea is itself a benign condition, however, it’s strong association with the development of glaucoma is important for physicians to be aware of in order to prevent future vision loss in these patients. If a patient is diagnosed with congenital ectropion uvea, regular tonometry evaluations are needed for their entire lifetime in order to monitor intraocular pressures. More extensive long-term follow-up is needed to establish which surgical intervention is the most successful in controlling intraocular pressures in patients with congenital ectropion uvea.
References:
- Harasymowycz, P. J., Papamatheakis, D. G., Eagle Jr, R. C. Wilson, R. P. (2006). Congenital ectropion uveae and glaucoma. Arch Ophthalmol, 124(2), 271-273. doi:10.1001/archopht.124.2.271
- Prenshaw, J. & Salim, S. (2013). Ectropion uveae and secondary glaucoma. Ophthalmic Pearls. Retrieved from https://www.aao.org/eyenet/article/ ectropion-uveae-secondary-glaucoma
- Seymenoglu, G. & Baser, E. (2011). Congenital iris ectropion associated with juvenile glaucoma. International Ophthalmology, 31(1), 33-38. doi: 10.1007/s10792-010-9388-6.
- Shifa, J. Z., Nkomazana, O., Bekele, N. A. , & Kassa, M. W. (2016). A young Botswana patient with congenital iris ectropion uvea. The Pan African Medical Journal, 25(42). doi: 10.11604/pamj.2016.25.42.10593
- Wang, G. M., Thuente, D., & Bohnsack, B. L. (2018). Angle closure glaucoma in congenital ectropion uvea. American Journal of Ophthalmology Case Reports, 10, 215-220. doi: 10.1016/j.ajoc.2018.03.009
Identifier: Moran_CORE_29713
Retinoblastoma
Home / Ophthalmic Pathology / Additional Resources
Title: Retinoblastoma
Author: Amber Jimenez
Photographer: Moran Eye Center photography
Date: 10/17/2019
Keywords/Main Subjects: tumor, tumor suppressor, leukocoria, RB1
Diagnosis: Retinoblastoma
Brief Description:
Retinoblastoma is an intraocular malignancy of the retinal cells resulting from a disruption of a tumor suppressor gene, called RB1. The disease often presents in children within the first two years of life. The most common symptom of retinoblastoma is leukocoria, or white pupil. One, or both eyes may be affected, and extent of the disease may vary, depending largely on the type of mutation being germline or somatic. Left untreated, retinoblastoma may have debilitating effects on vision and can be fatal. Prompt referral for management of the condition is recommended.
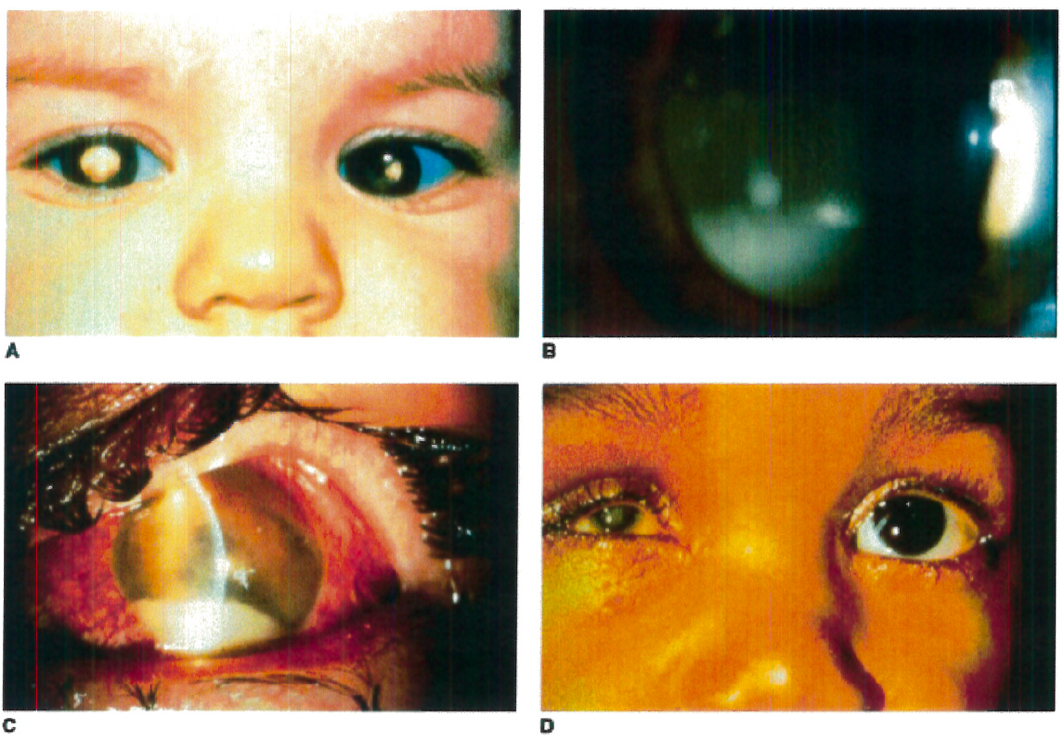
Figure 1. A: Heritable RB patient presenting with bilateral leukocoria, the most commonly reported presenting sign of this condition. B: RB cells have a propensity to detach from the primary tumor mass and spread throughout the eye, as illustrated by a patient with endophytic RB who developed marked tumor cells in the vitreous, resembling vitritis. C: Occasionally tumor necrosis may produce an inflammatory response associated with a red eye and conjunctival injection as seen in this patient. The patient also has a tumor hypopyon. D: Massive tumor necrosis in the phthisical eye of a patient with RB has resulted in marked inflammation involving the orbit, simulating an orbital cellulitis with lid swelling and pain on eye movement.
Taken from Figure 26.4 A-D, Hartnett, M.E. (2014) Pediatric Retina. Philadelphia, PA. Lippincott Williams & Wilkins, a Wolters Kluwer business.

Taken from Figure 26.5 B, Hartnett, M.E. (2014) Pediatric Retina. Philadelphia, PA. Lippincott Williams & Wilkins, a Wolters Kluwer business.
Age & Gender: The incidence of retinoblastoma is similar in males and females. Most cases are diagnosed at birth or early infancy, with an average age of diagnosis of bilateral disease at 12 months and 24 months for patients with unilateral disease.
Genotype: Both heritable (germline mutation) and nonheritable (somatic mutation) retinoblastoma arise from mutations in the retinoblastoma gene, RB1, on both alleles of chromosome 13q14.
Symptoms: The most common clinical presentation of retinoblastoma includes symptoms of leukocoria, white pupil, or strabismus in a child under 2 years of age. Other symptoms include nystagmus or a red inflamed eye.
Environmental Factors or Other Considerations: Germline mutations of RB1 have been found to occur on paternally derived chromosomes, suggesting that increased paternal age may have impact on the occurrence of retinoblastoma. No other environmental factors have been identified.
Treatment: Treatment options vary depending on the size and location of the tumor, extent of visual disturbance, optic nerve involvement, and presence of metastatic disease. Surgery, brachytherapy or radiation, chemotherapy, and laser therapy are common treatments.
Pathophysiology: The RB1 gene encodes the Rb protein which functions as a tumor suppressor by restricting progression of the cell cycle in dividing cells. Loss of function in both copies of RB1 causes cell cycle dysregulation, allowing for potentially cancerous cells to continue dividing. The Knudson “two-hit” model differentiates the heritable and nonheritable forms. Patients with the heritable form of retinoblastoma inherit a germline mutation of RB1 in all cells in the body, thus creating the “first hit.” A “second hit” occurs when the second allele of RB1 is mutated or silenced through epigenetic changes, affecting retinal cells. Patients with heritable retinoblastoma are at increased risk for bilateral and multifocal disease. Nonheritable retinoblastoma occurs with somatic mutations of both copies of the RB1 gene (both “first” and “second” hit) in a retinal cell. These patients present with unilateral and unifocal disease, and which usually occurs later in life.
Differential Diagnosis: The most common differential diagnoses include conditions associated with the most common presenting symptom of retinoblastoma, infantile leukocoria. Such conditions include congenital malformations such as persistent fetal vasculature, vascular changes such as Coat’s disease, and inflammatory diseases such as ocular tococariasis. A patient’s age, combined with a consistent clinical presentation, morphologic features, and family history, are key to differentiating these conditions from retinoblastoma.
References: Originally published in: Hartnett, Mary Elizabeth. Pediatric retina: medical and surgical approaches. 2nd Ed. Philadelphia: Lippincott Williams & Wilkins, 2005.
Abramson, D. H., Frank, C. M., Susman, M., Whalen, M. P., Dunkel, I. J., & Boyd, N. W. (1998). Presenting signs of retinoblastoma. The Journal of Pediatrics,132(3), 505-508. doi:10.1016/s0022-3476(98)70028-9
Abramson, D. H., Beaverson, K., Sangani, P., Vora, R. A., Lee, T. C., Hochberg, H. M., Kirszrot, J., Ranjithan, M. (2003). Screening for Retinoblastoma: Presenting Signs as Prognosticators of Patient and Ocular Survival. Pediatrics,112(6), 1248-1255. doi:10.1542/peds.112.6.1248
Balmer A, Munier F. Differential diagnosis of leukocoria and strabismus, first presenting signs of retinoblastoma. Clinical ophthalmology (Auckland, NZ). 2007;1(4):431-439.
Faculty Approval by: M. E. Hartnett, MD
Copyright ©2019. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
A Brief Overview of Keratoconus and its Topographical Findings
Home / External Disease and Cornea / Corneal Dystrophies and Ectasias
Title: A Brief Overview of Keratoconus and its Topographical Findings
Author: Zachary Mortensen, MS4, MBA
Photographer: James Gilman, CRA, FOPS
Date: August 24, 2018
Keywords/Main Subjects: keratoconus, cornea, corneal ectasia, corneal topography, collagen cross-linking
Diagnosis: Keratoconus
Summary of the Case: Progressive keratoconus in a young patient that is marked by progressive astigmatism, thinning and steepening of the cornea. The patient is monitored with corneal topography. He is an excellent candidate for corneal collagen cross-linking.
Case:
A 14-year-old boy with seasonal allergic rhinitis presents with a four-year history of keratoconus in both eyes that continues to progress. He has asymmetric astigmatism and has used prescription glasses in the past, but he now uses rigid gas permeable contact lenses. He has some optic nerve atrophy in the left eye due to a sledding accident trauma seven months ago that is now likely stable. He has no known family history of keratoconus or other eye diseases. He uses preservative free artificial tears as needed for eye dryness and oral antihistamines as needed for allergies.
Upon presentation to our clinic, his best corrected visual acuity with gas permeable contacts was 20/30-2 and 20/60-1 in the right and left eyes respectively. Slit Lamp examination of the anterior segment examination reveals the following image (Figure 1):

Figure 1 The cone-shape appearance of this cornea is characteristic of keratoconus but is not always evident on slit lamp examination. Image taken by Jim Gilman, University of Utah John A Moran Eye Center.
Central thinning, Vogt’s striae, and iron rings were appreciated on corneal examination in both eyes, and an apical scar seen in the left eye. The rest of the exam was unremarkable besides temporal sloping and some pallor of the left optic disc, likely due to the history of trauma.
Refraction:
Manifest Refraction:
| Sphere | Cylinder | Axis | |
| Right | -3.25 | +7.00 | 156 |
| Left | -2.75 | +6.00 | 018 |
Images:
Atlas Corneal Topography
Corneal topography is the gold standard for screening patients for keratoconus. Early screening for keratoconus is helpful because patients often look normal on slit lamp examination. It is also important for monitoring progression.1 Below is a Zeiss Atlas report (Figure 2) of the patient that has both an axial curvature map and a “Rings Image” which is a Placido disc-based topography. The warmer (redder) areas mark the areas of greater steepening.

Figure 2 Atlas topography of the patient with keratoconus at age 12. In this patient with keratoconus, the curvature map shows irregular astigmatism that is asymmetric in presentation. There is an area of excessive inferior, mid-peripheral steepening.
Calculating an inferior/superior (I-S) value can also be helpful when looking for keratoconus. The ratio of the average power differences between the inferior hemisphere and superior hemisphere on the cornea is the I-S value. A positive value indicates that the inferior cornea is steeper. An I-S value higher than 1.8 has been used by some as the cut-off point for clinical keratoconus.3 If the I-S value is calculated between 1.4 to 1.8, keratoconus can be suspected.3
Scheimpflug tomography (Pentacam)
The patient’s first Scheimpflug tomographic images (Oculus Pentacam) were taken at age 14 (Figure 3). The Pentacam shows several elevation maps. One of the significant advantages of the Pentacam is in its ability to show the posterior elevation changes.4 The elevation maps show an anterior and posterior elevation of the cornea relative to the best fit sphere (BFS). On average, normal anterior elevation change values are between 1 and 2μm, while keratoconic anterior elevation change values are can be 20 μm or greater.4 On average, normal posterior elevation change values are between 2 and 3 μm, while keratoconic posterior elevation change values can be 39-45 μm or greater.4

Figure 3 Pentacam of left eye at age 14. Note that the steepness is not necessarily where the corneal is thinnest (see Corneal Thickness and Relative Pachymetry in top and bottom middle). The cornea is most thin centrally, which is common in keratoconus. There is also elevation of anterior and posterior cornea paracentrally (see Elevation in right column).
Diagnosis
This patient has keratoconus in both eyes with characteristic progression. The patient has continued to progress when compared to old Atlas and Pentacam images, though manifest refraction has not largely changed. This patient’s keratoconus will likely continue to progress through pubescence and young adulthood.
Management
This patient is encouraged to avoid eye rubbing, which is an associated risk factor.5 It is important to help patients manage anything that can cause them to continue rubbing their eyes, such as prescribing steroids for those with atopic diseases.
When the patient was first seen at 10 years of age, he was managed by correcting visual acuity with spectacles. As he aged, the ectasia progressed, so he then started using rigid gas-permeable contact lenses.
There are also surgical management options that have been discussed with the patient. Intrastromal corneal ring segments are plastic inserts that are implanted into the cornea to flatten it. This can help with the visual acuity but does not stop progression.1 Corneal transplant (keratoplasty) is often used after contact lenses are no longer helpful.1 After discussion, we decided to delay keratoplasty until college-age years in an effort to achieve his best outcome since a higher rate of graft failure has been noted in pediatric penetrating keratoplasties.6
Collagen cross-linking is an intervention approved by the FDA in 2016 that slows the progression of keratoconus by strengthening bonds in the cornea. The procedure involves applying riboflavin (Vitamin B2) drops, then exposing it to ultraviolet light, and a photosensitizer. As it is relatively new in the United States, this procedure may be difficult for patients to acquire health insurance coverage. This patient is a good candidate for collagen cross-linking, so we have worked with him and his insurance company so that he can receive treatment.
Conclusion
Keratoconus, the most common ectatic disease of the cornea, is characterized by progressive central or paracentral thinning and steepening of the cornea such that the cornea takes on the shape of a “cone” (Figure 3). Early-stage keratoconus may look normal on slit lamp examination. Because of this, topography has become the gold standard for screening patients for keratoconus and other corneal ectasias. Topography and tomography are also useful for monitoring disease progression. Regular monitoring can permit early treatments such as corneal collagen cross-linking or corneal transplantation. While each patient should be evaluated individually, collagen cross-linking may be chosen in young patients with progressive keratoconus.
References:
- Wayman, L. L., Trobe, J., Wilterdink, J. L., (2018). Keratoconus. UpToDate. Waltham, MA: UpToDate Inc. http://www.uptodate.com (Accessed on August 23, 2018)
- Anderson, D. Understanding Corneal Topography (unknown date). Paraoptometric Resource Center. Retrieved from https://www.aoa.org/Documents/optometric-staff/Articles/Understanding-Corneal-Topography.pdf
- Cavas-Martínez, F., De la Cruz Sánchez, E., Martínez, J. N., Cañavate, F. F., & Fernández-Pacheco, D. G. (2016). Corneal topography in keratoconus: state of the art. Eye and Vision, 3(1), 5.
- Belin, M.W., Khachikian, S. S., Holladay, J. T., Tehrani, M. New Advances and Technology with Pentacam (2008). Highlights of Ophthalmology. Retrieved from https://www.oculus.de/uploads/media/oculus_low_res.pdf
- Sugar, J., & Macsai, M. S. (2012). What causes keratoconus?. Cornea, 31(6), 716-719.
- Bernfeld, E., Epley, K. D., Woodward, M. A. (2017). Pediatric penetrating keratoplasty. EyeWiki. Retrieved from http://eyewiki.aao.org/Pediatric_penetrating_keratoplasty
Faculty Approval by: Mark Mifflin, MD; Griffin Jardine, MD
Copyright statement: Copyright Author Name, ©2018. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Identifier: Moran_CORE_25586
A Cocooned ACIOL Secondary to UGH Syndrome
Home / Lens and Cataract / Complications of Cataract Surgery
Title: A Cocooned ACIOL Secondary to UGH Syndrome
Author: Tania Padilla Conde, MSIV, University of South Dakota Sanford School of Medicine
Photographer: Dr. Alan Crandall
Date: 7/9/18
Image or video:

Figure 1: Cocooned IOL of the right eye.
Keywords/Main Subjects: UGH syndrome, cocooned IOL, uveitis, glaucoma, hyphema
CORE Category:
Section 11 lens and cataractà Complications of cataract surgeryà UGH syndrome
http://morancore.utah.edu/section-11-lens-and-cataract/
Diagnosis: Inflamed cocoon anterior chamber IOL secondary to UGH syndrome, right eye
Description of Image:
This patient is an 85-year-old, African woman who presented with a chief complaint of worsening severe ocular pain in her right eye over the last several months. Her ocular history is significant for uveitis, elevated intraocular pressure (IOP), and cystoid macular edema (CME) of her right eye. Her ocular surgeries include cataract surgery in her right eye involving an anterior chamber intraocular lens (ACIOL) and a second cataract surgery in her left eye 8 years involving a posterior chamber intraocular lens (PCIOL). At presentation, she was on alphagan three times a day and timolol twice a day for her elevated IOP.
On exam, her visual acuity was OD count fingers at 3ft and OS 20/500. IOP by tonometry was OD 12 and OS 10. Pupils were non-reactive OU. Extraocular movements were full and ortho OU. Slit lamp examination OD showed benign melanosis, peripheral anterior synechiae (PAS) and a dislocated cocooned ACIOL. We were unable to get a clear view of the anterior chamber and the iris. We were also unable to get a clear view of the fundus on dilated exam. OS exam was unremarkable.
The patient was subsequently diagnosed with uveitis-glaucoma-hyphema (UGH) syndrome and underwent a removal of the ACIOL, anterior vitrectomy and synechiaelysis of the right eye. Due to severe inflammation, the decision was made to leave the patient aphakic with the possibility of an IOL implant in the future. The explanted IOL was sent to pathology and was found to be a one-piece with haptic PMMA anterior chamber Kelman multiflex-style IOL, Microscopic examination of the IOL showed several small fine granules of pigment on the haptics.
Discussion
UGH syndrome is a rare condition that classically presents with uveitis, glaucoma, and hyphema in the setting of an anterior chamber IOL. However, it can be diagnosed when one, two, or all three of these conditions are present in the setting of any IOL causing irritation of the iris or angle structures.1 Although it is traditionally seen as a complication of anterior chamber IOLs, single-piece acrylic IOLs or sulcus IOLs, cases with posterior chamber IOLs have also been reported.2 UGH syndrome is commonly characterized by chronic inflammation, CME, secondary iris neovascularization, recurrent hyphemas, and glaucomatous optic neuropathy leading to a loss of vision.3 The diagnosis is clinical, based on the history and physical exam, and can be supplemented by ultrasound biomicroscopy.4
UGH syndrome results from peripupillary contact of the iris with the lens optic/haptics leading to mechanical irritation and erosion of uveal structures. This chafing leads to a breakdown of the blood-aqueous barrier and subsequent release of pigment, red blood cells, protein, and white blood cells into the anterior chamber.5 The resultant inflammation causes uveitis. The release of the red blood cells can additionally cause a hyphema. The increase in IOP is a result of red and white blood cells in the anterior chamber blocking the trabecular meshwork and/or by the destruction of outflow structures. Electron microscopy of explanted IOLs often reveals coccoid-like structures on the haptic surface and melanosomes, likely from damaged iris pigment epithelial cells.6
Medical management can include topical steroids for uveitis, IOP lowering topical and systemic medications for intraocular hypertension, and topical steroids and cycloplegics for hyphema.6 The use of anti-VEGF therapy has been shown to induce the regression of iris neovascularization and inflammation in uveitic macular edema.7 The definitive treatment is often surgical intervention including IOL repositioning, explantation and/or exchange.
References:
- Cheng L, Fox AR, Kam JP, Alward WLM. Uveitis Glaucoma Hyphema (UGH) Syndrome. EyeRounds.org. posted October 3, 2017; Available from: http://EyeRounds.org/cases/257-UGH-syndrome.htm
- Zhang L, Hood CT, Vrabec JP, Cullen AL, Parrish EA, Moroi SE. Mechanisms for in-the-bag uveitis-glaucoma-hyphema syndrome. J Cataract Refract Surg 2014;40(3):490-492. https://www.ncbi.nlm.nih.gov/pubmed/24417893
- Crowell EL. Uveitis-Glaucoma-Hyphema Syndrome. EyeWiki. http://eyewiki.aao.org/ Uveitis-Glaucoma-Hyphema Syndrome. Accessed August 1, 2018.
- Lima BR, Pichi F, Hayden BC, Lowder CY. Ultrasound biomicroscopy in chronic pseudophakic ocular inflammation associated with misplaced intraocular lens haptics. Am J Ophthalmol. 2014 Apr;157(4):813–817. e1. doi: 10.1016/j.ajo.2013.12.025. https://www.ncbi.nlm.nih.gov/pubmed/24398393
- Chang DF, Masket S, Miller KM, Braga-Mele R, Little BC, Mamalis N, Oetting TA, Packer M. Complications of sulcus placement of single-piece acrylic intraocular lenses. Recommendations for backup IOL implantation following Posterior Capsule Rupture. J Cataract Refract Surg 2009;35:1445-58.
- Asaria RH, Salmon JF, Skinner AR, Ferguson DJ, McDonald B. Electron microscopy findings on an intraocular lens in the uveitis, glaucoma, hyphaema syndrome. Eye.1997;11(Pt 6):827–829. https://www.ncbi.nlm.nih.gov/pubmed/9537139
- Ellingson FT. The uveitis-glaucoma-hyphema syndrome associated with the Mark VIII anterior chamber lens implant. J Am Intraocul Implant Soc 1978;4(2):50-53. https://www.ncbi.nlm.nih.gov/pubmed/701165
- Rech L, Heckler L, Damji KF. Serial intracameral bevacizumab for uveitis glaucoma hyphaema syndrome: a case report. Can J Ophthalmol. 2014;49:e160–e162. https://www.ncbi.nlm.nih.gov/pubmed/25433757
Faculty Approval by: Dr. Alan Crandall and Dr. Griffin Jardine
Copyright statement: Copyright Tania Padilla Conde, ©2018. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
Disclosure (Financial or other): None
Identifier: Moran_CORE_25561
Ophthalmic Manifestations of Mucopolysaccharidosis Type IS (Scheie Syndrome)
Home / Retina and Vitreous / Retinal Degenerations Associated With Systemic Disease
Title: Ophthalmic Manifestations of Mucopolysaccharidosis Type IS (Scheie Syndrome)
Author: Shane Nau M.S., 4th Year Medical Student, University of Colorado School of Medicine
Photographer: Moran Eye Center Photography (Images 1-3), Lydia Sauer MD (Images 4-5)
Date: February 2016 (Images 1-3), June 2018 (Image 5)
Keywords/Main Subjects: Mucopolysaccharidosis; Lysosome Storage Disorder; Metabolic Disease; Corneal Dystrophy; Retinal Degeneration; Retinitis Pigmentosa; RP; Cystoid Macular Edema; CME; Fluoroscein Angiography; FA; Autofluorescence; AF; Fluoroscein Lifetime Imaging Ophthalmoscope; FLIO
Secondary CORE category:
External Disease and Cornea / Corneal Dystrophies and Ectasias
Pediatric Ophthalmology and Strabismus / Ocular Manifestations of Systemic Disease
Diagnosis: Mucopolysaccharidosis Type IS (Scheie Syndrome)
Brief Case Description:
HPI:
The patient is a 37-year-old female referred to retina clinic for evaluation of bilateral cystoid macular edema (CME) in early 2016. Upon presentation she endorsed blurry vision, 3 years of nyctalopia, and 10 years of floaters and scintillations. Her past ocular history is notable for bilateral corneal dystrophy of unknown etiology, which led to a penetrating keratoplasty (PKP) in her left eye. This procedure was complicated by pupillary-block glaucoma that later required cataract surgery. Despite a corneal transplant in the left eye, her vision remained worse in the left eye compared to the right. OCT imaging of her macula was obtained—revealing bilateral CME.
Her past medical history was significant for hand and foot contractures noted since early childhood, mitral valve stenosis noted in adolescence, severe arthritis, and an umbilical hernia that has since been repaired. She has mildly coarse facial features. She does not have a family history of ocular problems or autoimmune disease. She is allergic to Amoxicillin/Penicillin and presented taking both Ilevro (once daily) and Prednisolone (4X daily) eye drops in addition to 20 mg daily oral Prednisone daily.
Eye Exam: (see Image 1)
Vision: 20/60 cc OD and 20/100 cc OS
Pupils: 5>3, hazy view OD and 7>7, irregular OS
External: Normal OU
Slit Lamp: Diffuse corneal clouding OD, PKP OS clear with peripheral corneal clouding
Fundus Exam: Mild hyperemia of disc, CME, Mottled looking periphery OU
Workup:
CXR: No evidence of Tuberculosis or Sarcoidosis
ECG: Normal
FTA-ABS: Non-reactive
RPR: Non-reactive
ACE: 23 (N)
Lysozyme: <0.70 (N)
Retinal Autoantibodies: (+) for 23-kDA HSP27, 28-kDA, 46-kDA Enolase
MPS Urine Quant.: 12.5 (0-7.1)
MPSI Creatinine: 84 (0)
Total GAG Concentration: 11.5 (0-7.1)
Discussion:
Based on the clinical picture outlined above, it becomes clear that this patient presented with a rare combination of bilateral corneal clouding and retinopathy. Each of these respective findings have lengthy differentials. However, assuming these disease processes share a common etiology, the differential is dramatically narrowed. Using this clinical reasoning, we developed a high index of suspicion for more rare diseases such as the Mucopolysaccharidoses (MPS). Subsequently, a more direct and cost-effective workup was initiated. Quickly thereafter, the MPS labs confirmed the diagnosis of MPS Type IS (Scheie Syndrome) allowing the proper treatment regimen to be initiated.
Mucopolysaccharidosis Type IS (MPS IS, Scheie syndrome) is a type of lysosome storage disorder. Most of the mucopolysaccharidoses are mutations that are inherited in autosomal recessive fashion. With regards to Scheie syndrome, the mutation leads to a deficiency in the enzyme α-L-iduronidase, which is responsible for the degradation of glycosaminoglycans (GAG’s) within the body [1]. Without proper functioning of this GAG degradation process, chronic and progressive deposition of GAG’s throughout the body ensues. The eyes are no exception. According to Ashworth et. al., cytoplasmic membrane-bound vacuoles containing GAG’s are found in most ocular tissue (i.e. cornea, conjunctiva, lens epithelial cells, choroid, ciliary body, RPE, ganglion cells, and trabecular endothelium) in MPS IS patients. This extensive GAG deposition in ocular tissues causes structural and functional changes within the eye.
The following ophthalmic findings have been reported in Scheie Syndrome: proptosis, progressive corneal opacification, corneal edema, open angle glaucoma, pupillary- block glaucoma, cataracts, retinitis pigmentosa, and epiretinal membranes [2]. While the ophthalmic findings are essential to an ophthalmologist, it is contextually important to recognize the other clinical features associated with Scheie syndrome when making the diagnosis. According to Beck et al., the following clinical features of Scheie syndrome are ordered from most to least common: corneal clouding, joint contractures, cardiac valve abnormalities, hernias, carpal tunnel syndrome, and coarse facial features [3]. Each of these findings is found in the majority of Scheie syndrome patients. Perhaps more importantly, the patient in this case had each of these findings at some point in her first three decades of life. Had a provider picked up on her unique clinical history, it is possible she may have been diagnosed at a much earlier age.
Traditionally, ophthalmologic treatment of Scheie syndrome has focused on long-term management with conservatively-timed interventions (i.e. PKP for progressive corneal clouding). However, with the more recent advent of gene and enzyme replacement therapy, new options are becoming available for patients. The patient in this case was started on an experimental enzyme replacement therapy and her CME was tracked with serial OCT imaging. While anecdotal, her CME improved throughout treatment and acutely worsened when the study was ended.
Image/Video: Slit lamp (Image 1), OCT of macula (Image 2), Fluorescein Angiography (Image 3), Autofluorescence (Image 4), Fluorescein Lifetime Imaging Ophthalmoscopy (Image 5)
Description of Images:
Image 1: Slit lamp photo demonstrating diffuse corneal clouding of the right eye.

Image 2: OCT demonstrating cystoid macular edema (CME) with loss of the IS/OS junction surrounding the macula
Image 3: A late fluorescein angiogram photo of the left eye. There are hyper-fluorescent spots surrounding the macula indicating CME as well as significant vascular leakage in the mid-peripheral retina
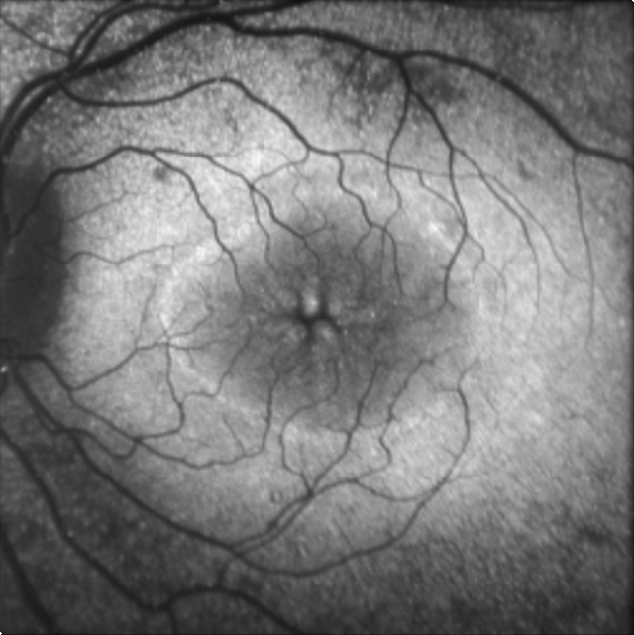
Image 4: Autofluorescence of the right eye showing a ring-shaped pattern of hyper- and hypo-autofluorescence surrounding the macula.
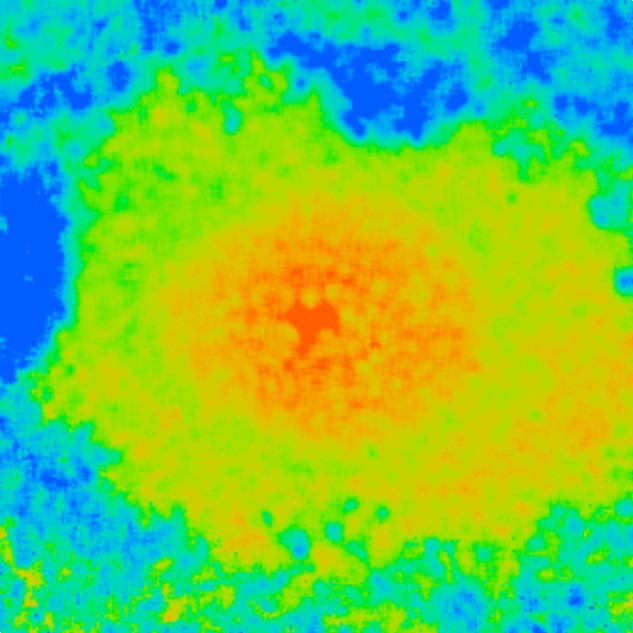
Image 5: Fluorescein Lifetime Imaging Ophthalmoscopy demonstrating a ring-shaped pattern of prolonged autofluorescence surrounding the macula.
Case Summary:
A 37-year-old patient presented with complaints of blurriness, nyctalopia, and worsened vision in her left eye despite a recent PKP (OS only) for non-specific bilateral corneal dystrophy. She was noted to have bilateral CME and loss of her IS/OS junction on OCT. Autofluorescence and Fluorescence Lifetime Imaging Ophthalmoscopy (FLIO) demonstrated an abnormal ring-shaped pattern around the macula similar to pattern seen with Retinitis Pigmentosa. Operating on the assumption that there was a common etiology for the corneal dystrophy and retinopathy, the diagnosis of mucopolysaccharidoses was suspected and later confirmed with labs. The patient successfully trialed an experimental enzyme replacement therapy and her CME worsened after the funding-based withdrawal of therapy. She has since undergone PKP in right eye as well. She is being followed every 6 months.
Format: Case Report
References:
- Thomas JA, Beck M, Clarke JTR, Cox GF. Childhood onset of Scheie syndrome, the attenuated form of mucopolysaccharidosis I. Journal of Inherited Metabolic Disorders. 2010; 33: 421-427.
- Ashworth J, et al. Mucopolysacharidoses and the Eye. Survey of Ophthalmology. 2006; 51: 1-17.
- Beck M, et al. The Natural History of MPS I: global perspectives from the MPS Registry. Genetics in Medicine. 2014; 16: 759-765.
- Andersen KM, Sauer L, Gensure RH, Hammer M, Bernstein PS. Characterization of retinitis pigmentosa using fluorescence lifetime imaging ophthalmoscopy (FLIO). TVST 2018; 7, 20.
- Lim, J. et al. Retinitis Pigmentosa. Eye Wiki. 23 May 2018, http://eyewiki.aao.org/Retinitis_Pigmentosa#Disease Accessed 20 June 2018
- Van C, Syed NA. Epithelial-Stromal and Stromal Corneal Dystrophies: A Clinicopathologic Review. Revision of [Birkholz ES, Syed NA, Wagoner, MD. Corneal Stromal Dystrophies: A Clinicopathologic Review. Aug. 17, 2009]; EyeRounds.org. August 20, 2015. Available from: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm
Faculty approval by: Akbar. Shakoor, MD
Footer:
- Copyright statement: Copyright Author Name, ©2016. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
Disclosures: None
Identifier: Moran_CORE_25548