


Commotio Retinae
Home / Retina and Vitreous / Posterior Segment Manifestations of Trauma
Title: Commotio Retinae
Author: Joanna Gorka, BS
Date: 8/5/24
Keywords/Main Subjects: commotio retinae, blunt globe trauma, retinopathy, OCT
Diagnosis: Commotio Retinae
Description of Case:
Introduction
Commotio retinae, derived from the latin word commōtus referring to ‘disturbance’ or ‘agitation’ is a traumatic retinopathy secondary to direct or indirect trauma to the globe.
Case
A 42-year-old male presented with diminished vision and sharp pain in the left eye one day after blunt trauma via metal hook of a bungee cord. He reported a complete loss of vision in the eye immediately following the injury which shortly after improved to blurriness with diminished superior visual field. On evaluation, visual acuity was 20/40. No corneal lacerations were appreciated. Fundus exam revealed commotio of the temporal/inferior retina, significant retinal mottling, and scattered pre-retinal hemorrhage.
One-week post-injury, the patient returned to clinic with worsened vision, 20/60. OCT was notable for slight hyperreflectivity to the outer retina consistent with commotio. 2 months later, the patient’s visual acuity recovered to 20/20. Post-traumatic peripheral retinal pigmentary changes were present 4 months post-injury.
Images:
Day 0: 20/40. Multilayered hemorrhage inferior/temporal and retinal whitening consistent with commotio retinae.
2 weeks: 20/60. Slight hyperreflectivity to outer retina consistent with commotio.
2 months: 20/20. Temporal/inferotemporal pigmentary changes with subretinal hemorrhage. Temporal small area of localized elevation.
4 months: 20/20. Temporal/inferotemporal pigmentary changes with flattened old blood inferiorly. Temporal small area of localized vitreous fibrosis over retina.
Summary of the Case: Commotio retinae is a self-limited retinopathy secondary to blunt globe trauma that is often characterized by retinal whitening.1 OCT may show increased reflectivity and thickness of the ellipsoid zone. Ahn et al. developed a 4-step grading system for prognosis based on OCT findings. OCT findings in this patient case are consistent with Grade 1 findings which are associated with the highest likelihood of visual and anatomic recovery.2 Patients with commotio retinae should be followed with serial examinations and monitored for the development of macular holes.3 There is no indicated medical treatment for commotio retinae, and most patients experience full recovery.4
References: BLANCH, R.J., UNDERSTANDING AND PREVENTING VISUAL LOSS IN COMMOTIO RETINAE, in College of Medical and Dental Sciences. 2014, University of Birmingham: Birmingham, UK. p. 581.
- Ahn, S.J., et al., Optical coherence tomography morphologic grading of macular commotio retinae and its association with anatomic and visual outcomes. Am J Ophthalmol, 2013. 156(5): p. 994-1001.e1.
- David Browning, M.P. What You Should Know About Blunt Trauma to the Eye: Commotio Retinae, Hyphema, Lens Dislocation, Vitreous Hemorrhage, Retinal Breaks, and Early and Late Glaucoma. [cited 2016 May 30th, 2016]; Available from: http://www.retinareference.com/diseases/9c563c5794aacd24/documents/9c563c5794/document.pdf.
- Blanch RJ, Good PA, Shah P, Bishop JR, Logan A, Scott RA. Visual outcomes after blunt ocular trauma. Ophthalmology. 2013 Aug;120(8):1588-91. doi: 10.1016/j.ophtha.2013.01.009. Epub 2013 Apr 22. PMID: 23618228.
Faculty Approval by: Paul S. Bernstein, MD, PhD
Copyright statement: Joanna Gorka, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Visual Impairment in an Aging Population
Basic Ophthalmology Review / Additional Resources
Title: Visual Impairment in an Aging Population
Authors: Michael Jensen, MD., Brian Stagg MD
Date: 9/5/2024
Keywords/Main Subjects: Aging, visual impairment
Summary: Visual impairment and eye disease disproportionately affect older adults ≥65 years. With the growing, aging population, the prevalence of VI is expected to double by 2050. Projected shortages of ophthalmologists indicate the work force will likely be inadequate to meet the growing demand. VI prevalence and access to care significantly impacts public health planning, Medicare disbursements, ocular disease, and common co-morbidities such as dementia and depression.
Introduction
The prevalence of vision impairment (VI) and visual dysfunction disproportionately affects older adults.1 Recent studies show that 1-in-4 adults over age 71 have visual impairment, defined as VA >20/40.2,3 Additionally, 10% have visually significant reduction in contrast sensitivity, and another 10% have distance vision impairment. The prevalence of VI increases to 37.9% of adults 85-89 and 46% in adults ≥90 years.3 By 2050, it is estimated there will be 61million people (about twice the population of Texas) legally blind worldwide, and 474 million with moderate to severe VI, the majority of whom will be over age 65.11
Currently in the US, there are an estimated 60 million adults over 65 years, however, the number of older adults is expected to increase to 78.3 million by 2040 and 82 million by 2050.4 This amounts to an increase in total share of the population from 17% to 23%. Over that same time, life expectancy will increase, from 73.6 years to 78.2 years, which will increase the proportion of adults in the 8th and 9th decade of life which is when vision impairment tends to be more severe and disabling.4 An expanding, aging population is the primary driver of the growing burden of VI. The resulting demand for eye health services is expected to increase accordingly and current projections forecast a significant shortage of ophthalmologists by the year 2035.5 Of 38 specialties, ophthalmology is projected to rank 37th in terms of adequate physician coverage.5
By the Numbers
In 2024, the Department of Human and Health Services and Centers for Medicare and Medicaid Services (CMS) released physician-level Medicare disbursement data.6 The report contains data from 2012 detailing the quantity and type of health care provided under the Medicare Part B fee-for-service (FFS) program from all 50 states, Washington DC, and Puerto Rico. The report found that ophthalmology as a specialty received the second highest aggregate Medicare payments at just over 5 billion dollars. Most of the payments were to ambulatory surgical centers for cataract surgery and anti-vascular endothelial growth factor (anti-VEGF) injections for treatment of neovascular age-related macular degeneration (ARMD) and other retinal diseases. Ophthalmology’s place in second is not surprising given historical trends dating back to the 1980’s.7 A similar study published in 1992 found that ophthalmology was the largest recipient of Medicare specialty disbursements.7
Cataracts and ARMD are among the most common eye diseases in older adults. In 2010, an estimated 50% of adults ≥75 years had visually significant cataracts. NIH estimates in 2050 there will be 50.2 million people in the United States alone that will have visually significant cataracts.8 In addition to cataracts, 20% of adults ≥75 years have early AMD. The prevalence increases to 30% by age 85. Late-stage AMD is relatively rare given modern therapeutics, but prevalence increases dramatically with age, from 3-4% at age 75, 10% at age 80, up to15% at age 90.9
Glaucoma is another ocular disease more common in older adults. Glaucoma prevalence ranges 4-8% for adults 65-75, and 7-12% over 80 years.10,11 Glaucoma is the leading cause of irreversible blindness and typically has an insidious onset over many years where the patient has no perception of their reducing vision. Topical medications in the form of eye drops are the most common 1st line treatment. Application of eye drops can be difficult for older adults due to physical limitations secondary to loss of the natural lordosis of the spine, poor vision, reduced coordination, and overall medication burden. However, non-adherence rates by age vary across studies with some studies showing reduced adherence, increased adherence, or no difference in adherence based on age.12-14 Older adults do tend to report higher rates of difficulty with eye drops and are more likely to refill prescriptions before the refill date due to wastage.15
A population-based study from 2021 showed that prevalence of diabetic retinopathy (DR) to be 30% in adults with diabetes age ≥75, of which 8-10% have vision threatening DR.16 These results are similar to a meta-analysis in 2004 which found the prevalence of DR to range between 10-40% and vision threatening DR to range between 2-10% in adults ≥75 years.17 With rates of diabetes projected to increase, there may be a resulting increase in the prevalence of DR and vision threatening DR.18
Dry eye occurs in 5-30% of adults over age 65 and prevalences increases with age. With aging there are changes in tear quality, increased tear film evaporation, neurodegenerative disease such as Parkinson’s disease which decrease tear production, and increased inflammation and oxidative stress which also disrupts tear film production.19 Dry eye is a risk factor many other ocular diseases, especially corneal disease. Uncorrected refractive error is another common cause of VI. 17.8-23.60% of adults ages 65-79 have ≥ +3 D of hyperopia. Myopia, however, is inversely correlated with age.20
There are numerous other causes of VI that are rare in the population over all but more common in older adults. For example, the median age of diagnosis for uveal melanoma is approximately 62 years and the peak range for diagnosis is between 70 and 79 years.21 Retinal detachment, optic neuropathy, and dermatochalasis are other examples of ocular disease more prevalent in older adults.22-24
Multi-Morbidity
Visual impairment has significant consequences on the health of older adults apart from their vision. Cardiovascular disease, diabetes, hypertension, hearing impairment, COPD, kidney disease, and stroke are more common in adults >65 years with vision impairment compared to age matched, visually healthy controls.25,26 Poor vision also effects how a person with chronic disease perceives their health. Self-reporting studies show that patients with VI report their health as worse compared to disease-matched controls without VI.26 A recent meta-analysis of 28 studies found that the hazard of all-cause mortality was higher in people with moderate to severe VI compared to those with normal vision or mild VI.27
A recently published nationally representative study with objective visual measurements found that the prevalence of falls is associated with decreased contrast sensitivity but not with near or far distance visual acuity.28 Currently, contrast sensitivity screening is not included in the NIH Toolbox of recommended visual screening tests, or the Center of Disease Control’s (CDC) Stopping Elderly Accidents, Deaths, & Injuries (STEADI) algorithm.29,30 More research needs to be done to assess the utility of regular contrast sensitivity screening by general practitioners and geriatricians.
Untreated poor vision has been identified as a contributing factor to dementia later in life. Rogers and colleagues found that the risk of developing dementia was 56% lower in participants who received an indicated eye procedure than those without and that the risk of dementia decreased by 64% in those with at least one visit to an ophthalmologist.31 Another study using data from the 2018 Health and Retirement Study estimates that more than 100,000 cases of dementia could have been prevented through healthy vision.32 VI is reported to be as high as 74% in patients with cognitive decline.33 VI and visual symptoms lead to a positive feedback cycle of more frequent and severe neuropsychiatric symptoms, increased hospital services, decreased social interaction, and increased conflict and exhaustion for caregivers.33
Depression and depressive symptoms are also more common in older adults with sensory difficulty, including VI, compared to age-matched controls.1834 In a recent study, visual difficulty was found to have a hazard ratio of 1.25 for incidence of depressive symptoms, compared to those without sensory difficulty. The cause is likely multi-factorial including increased withdrawal from social activity, loneliness, inability to participate in activities that promote wellness and wellbeing, discrimination and stigma.34,35
Conclusion
Visual impairment and eye disease disproportionately affect older adults ≥65 years. With the growing, aging population, the prevalence of VI is expected to double by 2050. Projected shortages of ophthalmologists indicate the work force will likely be inadequate to meet the growing demand. VI prevalence and access to care significantly impacts public health planning, Medicare disbursements, ocular disease, and common co-morbidities such as dementia and depression.
References
- Bourne R, Steinmetz JD, Flaxman S, et al. Trends in prevalence of blindness and distance and near vision impairment over 30 years: an analysis for the Global Burden of Disease Study. Lancet Glob Health. 2021;9(2):e130-e143. doi:10.1016/S2214-109X(20)30425-3
- ICD-11 for Mortality and Morbidity Statistics 9D90 Vision impairment including blindness. Accessed August 12, 2024. https://icd.who.int/browse/2024-01/mms/en#1103667651
- Killeen OJ, De Lott LB, Zhou Y, et al. Population Prevalence of Vision Impairment in US Adults 71 Years and Older. JAMA Ophthalmol. 2023;141(2):197. doi:10.1001/jamaophthalmol.2022.5840
- 2023 National Population Projections Tables: Main Series. Accessed August 12, 2024. https://www.census.gov/data/tables/2023/demo/popproj/2023-summary-tables.html
- Berkowitz ST, Finn AP, Parikh R, Kuriyan AE, Patel S. Ophthalmology Workforce Projections in the United States, 2020 to 2035. Ophthalmology. 2024;131(2):133-139. doi:10.1016/j.ophtha.2023.09.018
- HHS Releases Physician-Level Medicare Data. Accessed August 13, 2024. https://www.cms.gov/newsroom/fact-sheets/hhs-releases-physician-level-medicare-data
- Frenkel M. Ophthalmology Is the Single Largest Recipient of Medicare Specialty Disbursements. Archives of Ophthalmology. 1992;110(2):168. doi:10.1001/archopht.1992.01080140018004
- Cataract Tables. Accessed August 12, 2024. https://www.nei.nih.gov/learn-about-eye-health/eye-health-data-and-statistics/cataract-data-and-statistics/cataract-tables
- Rein DB, Wittenborn JS, Burke-Conte Z, et al. Prevalence of Age-Related Macular Degeneration in the US in 2019. JAMA Ophthalmol. 2022;140(12):1202. doi:10.1001/jamaophthalmol.2022.4401
- Rudnicka AR, Mt-Isa S, Owen CG, Cook DG, Ashby D. Variations in Primary Open-Angle Glaucoma Prevalence by Age, Gender, and Race: A Bayesian Meta-Analysis. Investigative Opthalmology & Visual Science. 2006;47(10):4254. doi:10.1167/iovs.06-0299
- Zhang N, Wang J, Li Y, Jiang B. Prevalence of primary open angle glaucoma in the last 20 years: a meta-analysis and systematic review. Sci Rep. 2021;11(1):13762. doi:10.1038/s41598-021-92971-w
- Wolfram C, Stahlberg E, Pfeiffer N. Patient-Reported Nonadherence with Glaucoma Therapy. Journal of Ocular Pharmacology and Therapeutics. 2019;35(4):223-228. doi:10.1089/jop.2018.0134
- OLTHOFF C, SCHOUTEN J, VANDEBORNE B, WEBERS C. Noncompliance with Ocular Hypotensive Treatment in Patients with Glaucoma or Ocular HypertensionAn Evidence-Based Review. Ophthalmology. 2005;112(6):953-961.e7. doi:10.1016/j.ophtha.2004.12.035
- Tse AP, Shah M, Jamal N, Shaikh A. Glaucoma treatment adherence at a United Kingdom general practice. Eye. 2016;30(8):1118-1122. doi:10.1038/eye.2016.103
- Friedman DS, Okeke CO, Jampel HD, et al. Risk Factors for Poor Adherence to Eyedrops in Electronically Monitored Patients with Glaucoma. Ophthalmology. 2009;116(6):1097-1105. doi:10.1016/j.ophtha.2009.01.021
- Lundeen EA, Burke-Conte Z, Rein DB, et al. Prevalence of Diabetic Retinopathy in the US in 2021. JAMA Ophthalmol. 2023;141(8):747. doi:10.1001/jamaophthalmol.2023.2289
- The Eye Diseases Prevalence Research Group. The Prevalence of Diabetic Retinopathy Among Adults in the United States. Archives of Ophthalmology. 2004;122(4):552. doi:10.1001/archopht.122.4.552
- Ong KL, Stafford LK, McLaughlin SA, et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021. The Lancet. 2023;402(10397):203-234. doi:10.1016/S0140-6736(23)01301-
- Sharma A, Hindman HB. Aging: A Predisposition to Dry Eyes. J Ophthalmol. 2014;2014:1-8. doi:10.1155/2014/781683
- Eye Diseases Prevalence ResearchGroup. The Prevalence of Refractive Errors Among Adults in the United States,Western Europe, and Australia. Archives of Ophthalmology. 2004;122(4):495. doi:10.1001/archopht.122.4.495
- Krantz BA, Dave N, Komatsubara KM, Marr BP, Carvajal RD. Uveal melanoma: epidemiology, etiology, and treatment of primary disease. Clinical Ophthalmology. 2017;Volume 11:279-289. doi:10.2147/OPTH.S89591
- Jacobs LC, Liu F, Bleyen I, et al. Intrinsic and Extrinsic Risk Factors for Sagging Eyelids. JAMA Dermatol. 2014;150(8):836. doi:10.1001/jamadermatol.2014.27
- Saraf SS, Lacy M, Hunt MS, et al. Demographics and Seasonality of Retinal Detachment, Retinal Breaks, and Posterior Vitreous Detachment from the Intelligent Research in Sight Registry. Ophthalmology Science. 2022;2(2):100145. doi:10.1016/j.xops.2022.100145
- Hayreh SS. Ischemic optic neuropathy. Prog Retin Eye Res. 2009;28(1):34-62. doi:10.1016/j.preteyeres.2008.11.002
- Vision Impairment and Chronic Health Conditions. Accessed August 12, 2024. https://www.cdc.gov/vision-health/php/chronic-conditions-vision/index.html
- Crews JE, Chou CF, Sekar S, Saaddine JB. The Prevalence of Chronic Conditions and Poor Health Among People With and Without Vision Impairment, Aged ≥65 Years, 2010–2014. Am J Ophthalmol. 2017;182:18-30. doi:10.1016/j.ajo.2017.06.038
- Ehrlich JR, Ramke J, Macleod D, et al. Association between vision impairment and mortality: a systematic review and meta-analysis. Lancet Glob Health. 2021;9(4):e418-e430. doi:10.1016/S2214-109X(20)30549-0
- Jin H, Zhou Y, Stagg BC, Ehrlich JR. Association between vision impairment and increased prevalence of falls in older US adults. J Am Geriatr Soc. 2024;72(5):1373-1383. doi:10.1111/jgs.18879
- Varma R, McKean-Cowdin R, Vitale S, Slotkin J, Hays RD. Vision assessment using the NIH Toolbox. Neurology. 2013;80(11_supplement_3). doi:10.1212/WNL.0b013e3182876e0a
- STEADI – Older Adult Fall Prevention Algorithm. Accessed August 15, 2024. https://www.cdc.gov/steadi/index.html
- Rogers MAM, Langa KM. Untreated Poor Vision: A Contributing Factor to Late-Life Dementia. Am J Epidemiol. 2010;171(6):728-735. doi:10.1093/aje/kwp453
- Ehrlich JR, Goldstein J, Swenor BK, Whitson H, Langa KM, Veliz P. Addition of Vision Impairment to a Life-Course Model of Potentially Modifiable Dementia Risk Factors in the US. JAMA Neurol. 2022;79(6):623. doi:10.1001/jamaneurol.2022.0723
- Zhang W, Roberts T V., Poulos CJ, Stanaway FF. Prevalence of visual impairment in older people living with dementia and its impact: a scoping review. BMC Geriatr. 2023;23(1):63. doi:10.1186/s12877-022-03581-8
- Killeen OJ, Xiang X, Powell D, et al. Longitudinal Associations of Self-Reported Visual, Hearing, and Dual Sensory Difficulties With Symptoms of Depression Among Older Adults in the United States. Front Neurosci. 2022;16. doi:10.3389/fnins.2022.786244
- Shakarchi AF, Assi L, Ehrlich JR, Deal JA, Reed NS, Swenor BK. Dual Sensory Impairment and Perceived Everyday Discrimination in the United States. JAMA Ophthalmol. 2020;138(12):1227. doi:10.1001/jamaophthalmol.2020.3982
Faculty Approval by: Brain Stagg
Copyright statement: Michael Jensen, Brian Stagg, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Congenital Rubella Retinopathy
Home / Retina and Vitreous / Congenital and Developmental Abnormalities /
Title: Congenital Rubella Retinopathy
Authors: Alina Husain, MSIV, Weill Cornell Medical College; Elizabeth Fairless, MD, University of Utah
Date: 08/28/2024
Keywords/Main Subjects: Congenital rubella retinopathy, congenital rubella syndrome, pigmentary retinopathy
Diagnosis: Congenital rubella retinopathy
Images:
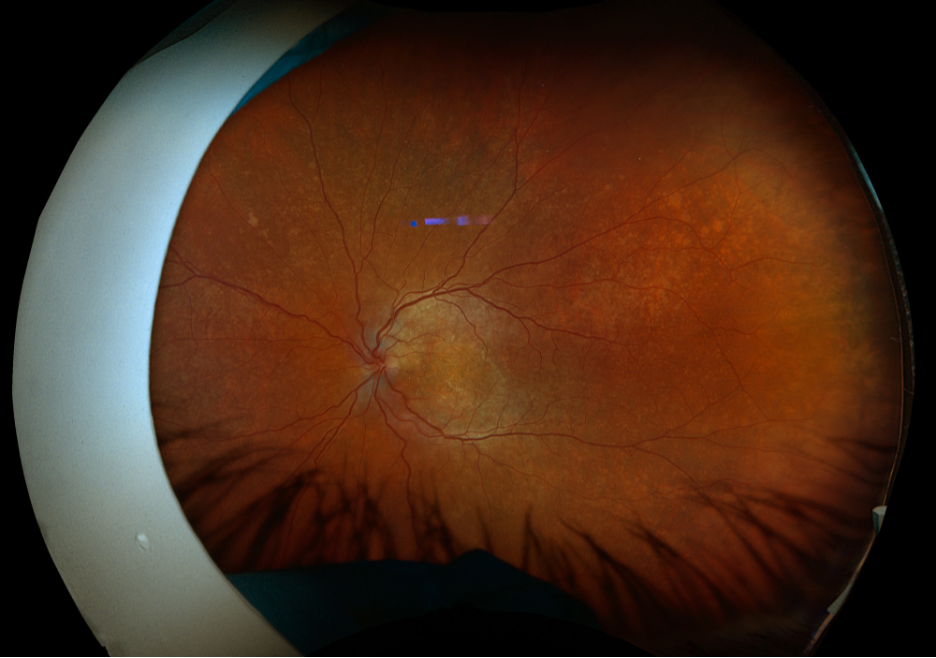
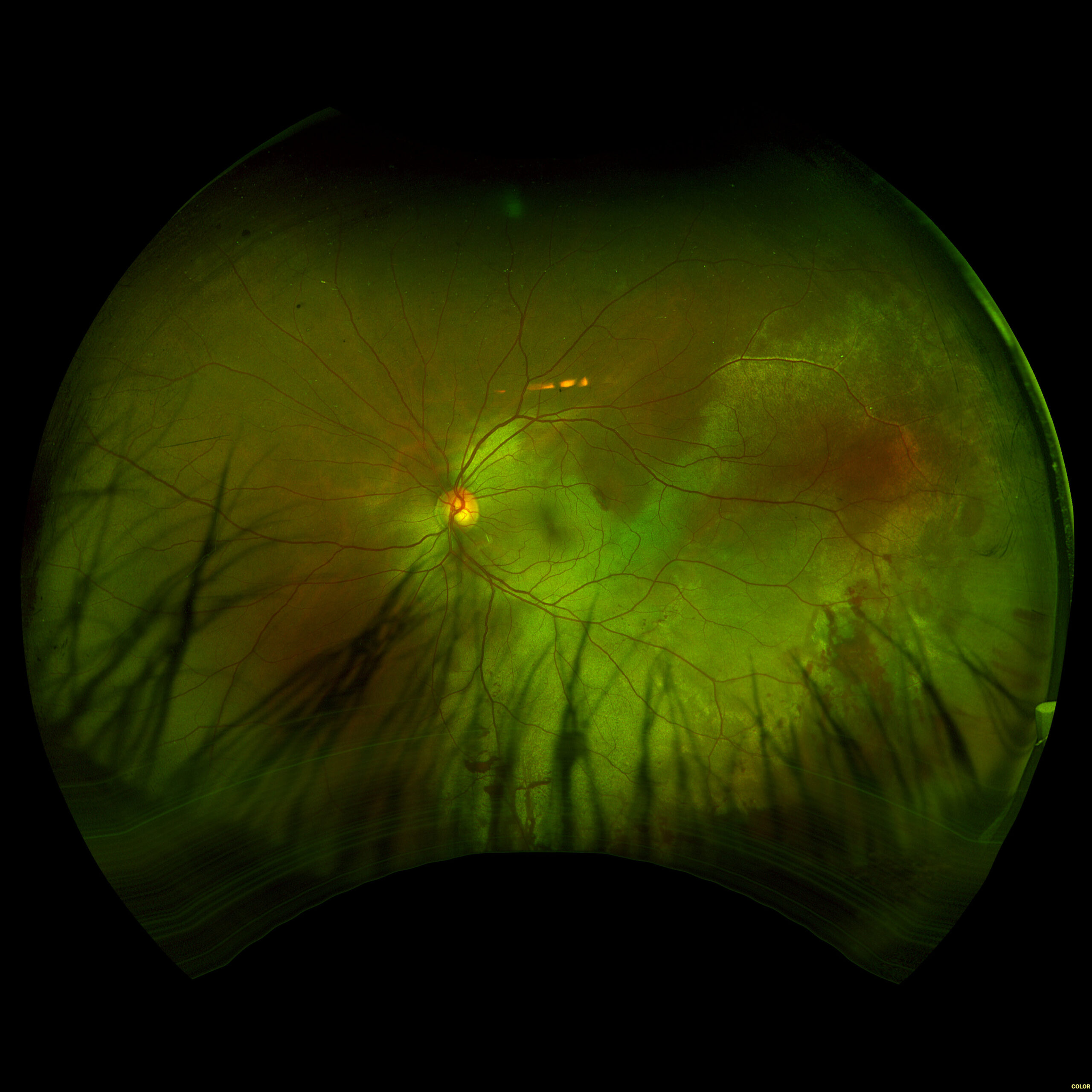
Figure 1. Fundus imaging of the left eye, demonstrating diffuse speckled pigmentary changes in the macula and periphery.
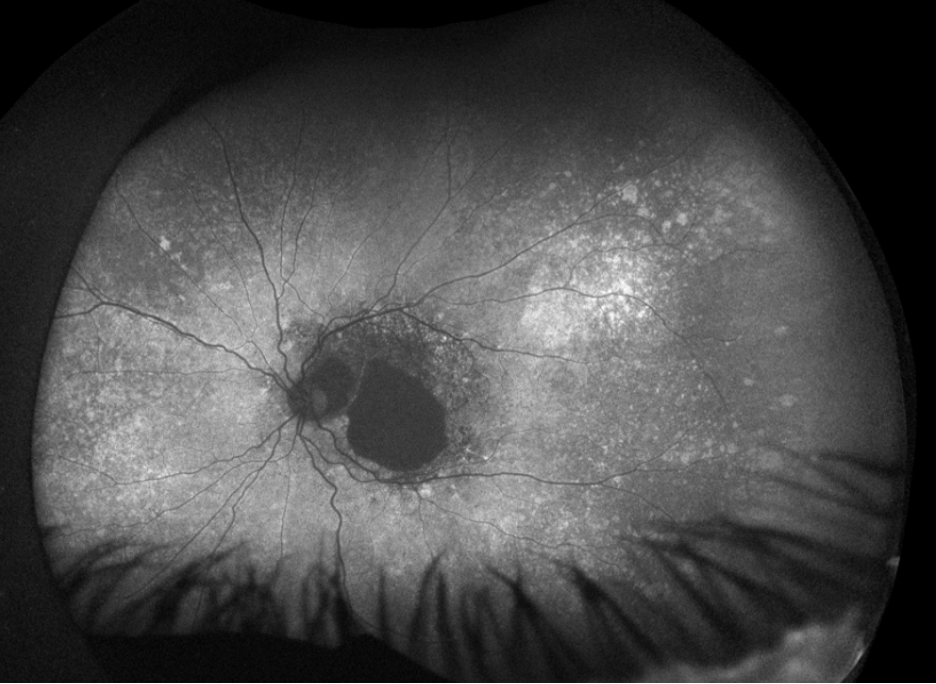
Figure 2. Fundus autofluorescence of the left eye, demonstrating hypoautofluorescence in the posterior pole, a well-defined area of hypoautofluorescence corresponding to macular atrophy, and mottled hypoautofluorescent and hyperautofluorescent lesions in the periphery.
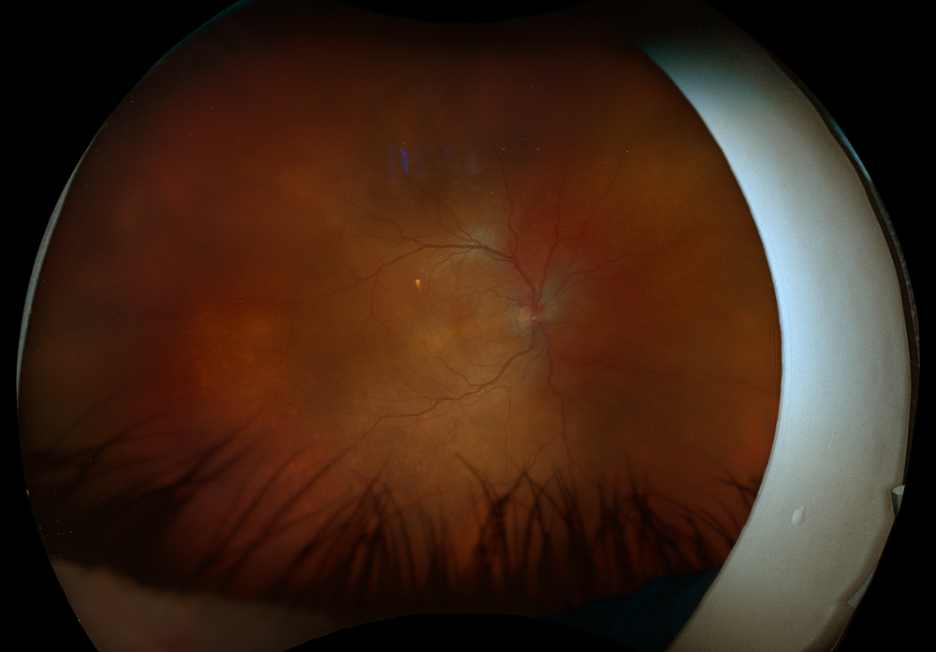
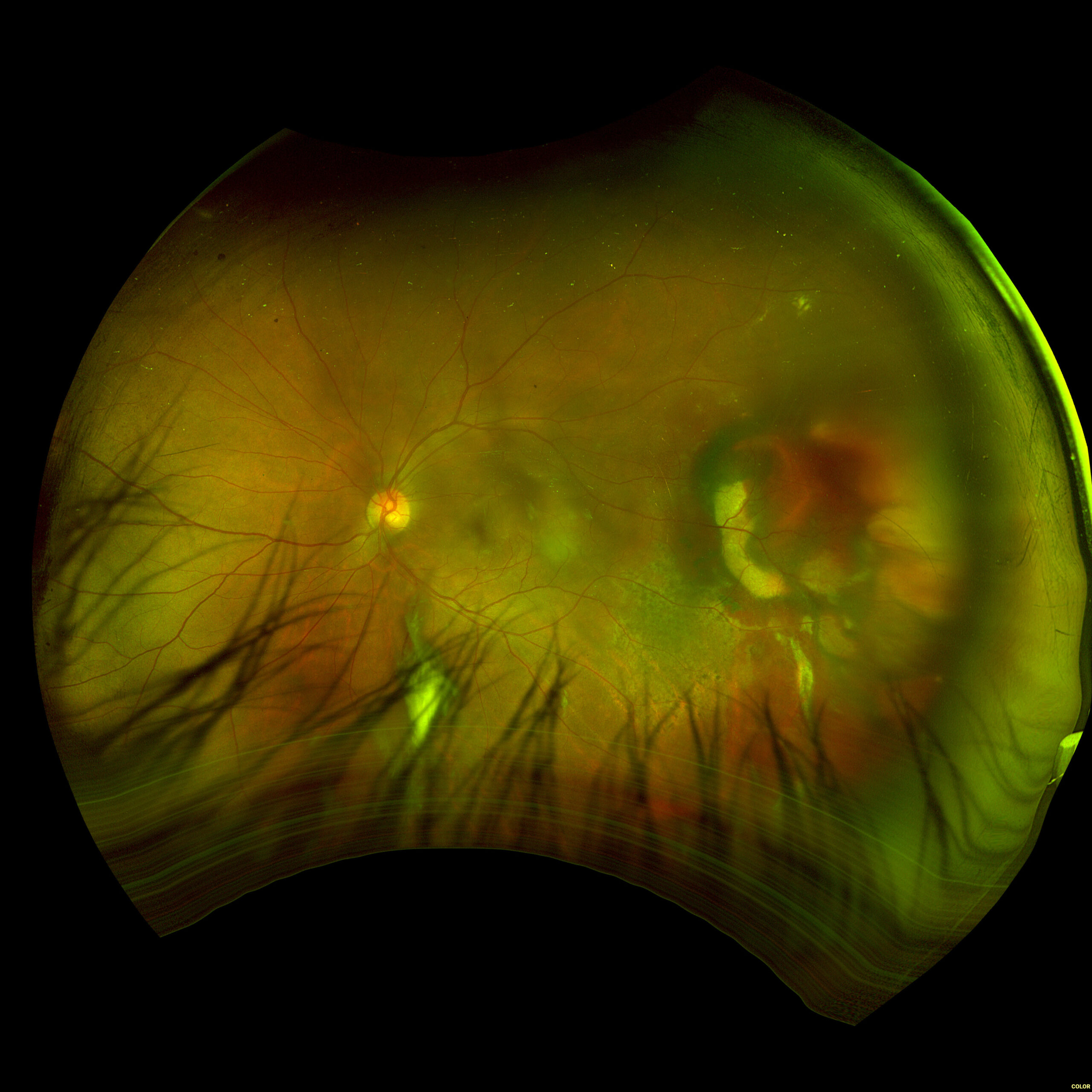
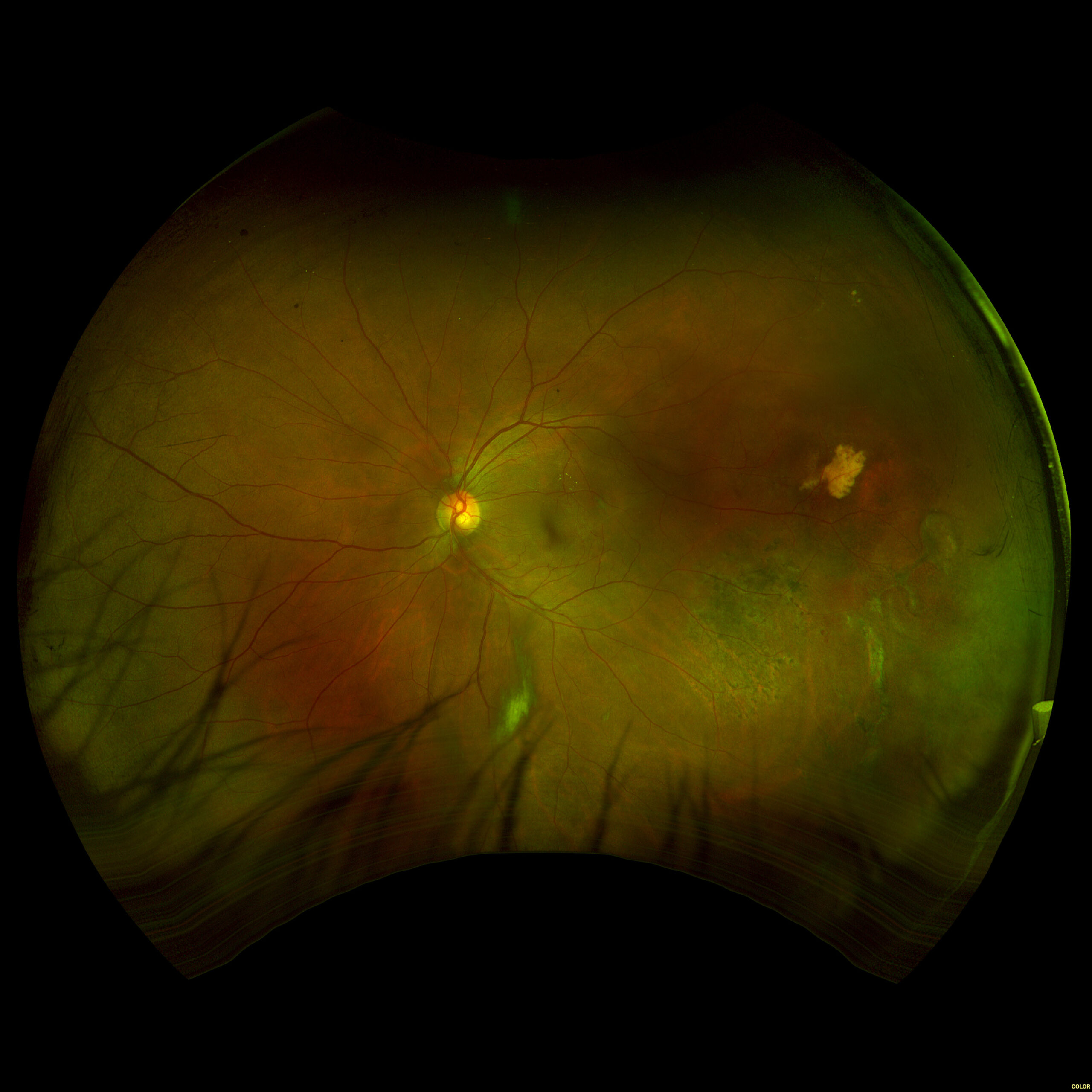
Figure 3. Fundus imaging of the right eye, demonstrating diffuse speckled pigmentary changes in the macula and periphery.
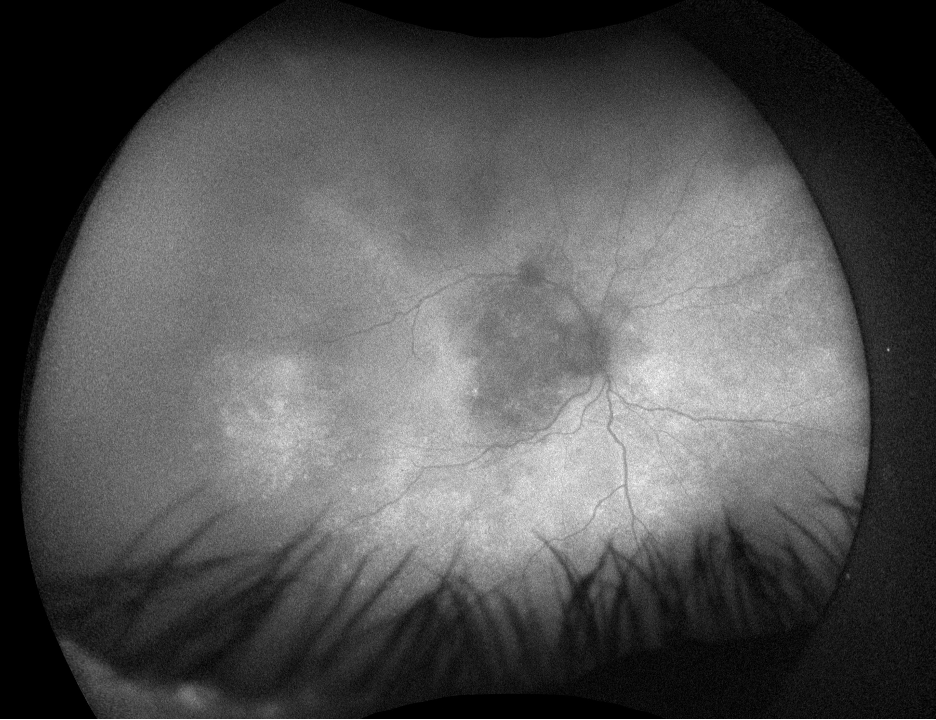
Figure 4. Fundus autofluorescence of the right eye, demonstrating hypoautofluorescence in the posterior pole and mottled hypoautofluorescent and hyperautofluorescent lesions in the periphery.
Description of Images:
Congenital rubella syndrome (CRS) is an uncommon syndrome caused by maternal infection with rubella during the first trimester of pregnancy. The incidence of CRS has decreased dramatically since the adoption of the rubella-containing vaccine, with only 1,527 cases of CRS reported worldwide in 2022.1 The pathophysiology of CRS is multifactorial and may be in part due to non-inflammatory necrosis of the chorionic epithelium and endothelial cells, disruption of intracellular actin assembly, and cytokine upregulation in infected cells.2 Non-ocular manifestations of CRS include congenital heart defects, sensorineural hearing loss, neurologic abnormalities, hematologic abnormalities, low birth weight, and delayed-onset thyroid disease and insulin-dependent diabetes,3,4 while ocular manifestations of CRS include cataracts, microphthalmos, pigmentary retinopathy, strabismus, glaucoma, and chorioretinitis.3,5 Currently, there is no cure for CRS, though some features of the condition can be treated or managed with supportive care.
The pigmentary retinopathy found in CRS can be unilateral or bilateral and has often been described as having a “salt-and-pepper” appearance given the presence of the hypo- and hyper-pigmented lesions often found in this condition; however, this appearance can be highly variable.6,7 The pigmentary changes associated with CRS are due to necrosis and atrophy of the retinal pigment epithelium caused by the rubella virus.8 Congenital rubella retinopathy can be progressive or non-progressive, and visual acuity and electroretinography is usually unaffected.7,9 Rarely, the retinopathy may cause subretinal neovascularization, which can lead to vision loss and disciform maculopathy.8,9 Funduscopic imaging typically demonstrates retinal pigmentary changes, with autofluorescence demonstrating stippled autofluorescence.7,10
These color and autofluorescence fundus photographs are from the eyes of a 56-year-old male with a past medical history notable for maternal rubella infection during pregnancy, congenital deafness, and autoimmune hemolytic anemia. He had a past ocular history of pigmentary retinopathy in both eyes, amblyopia in his left eye, and recent onset of recurrent non-granulomatous anterior uveitis in his right eye. He had a best-corrected visual acuity of 20/40 OD, 20/150 eccentric OS. Dilated funduscopic examination demonstrated a flat macula and diffuse speckled pigmentary changes in the macula and periphery of both eyes, with greater pigmentary changes in the left eye. The left eye additionally had a dense central area of retinal pigment epithelium and retinal atrophy, concordant with his poor visual acuity. See Figures 1 and 2.
Fundus autofluorescence demonstrates hypoautofluorescence throughout the posterior pole in both eyes, with a dense well-defined area of hypoautofluorescence in the left eye corresponding with the area of macular atrophy. Additionally, mottled hypoautofluorescent and hyperautofluorescent lesions are seen in the periphery in both eyes. See Figures 3 and 4.
These findings are consistent with congenital rubella retinopathy secondary to CRS.
References:
- Ou A, Zimmerman L, Alexander J, Crowcroft N, O’Connor P, Knapp J. Progress Toward Rubella and Congenital Rubella Syndrome Elimination — Worldwide, 2012–2022. Centers for Disease Control and Prevention; 2024:162-167. Accessed August 25, 2024. https://www.cdc.gov/mmwr/volumes/73/wr/pdfs/mm7308a2-H.pdf
- Bouthry E, Picone O, Hamdi G, Grangeot-Keros L, Ayoubi JM, Vauloup-Fellous C. Rubella and pregnancy: diagnosis, management and outcomes. Prenat Diagn. 2014;34(13):1246-1253. doi:10.1002/pd.4467
- Kaushik A, Verma S, Kumar P. Congenital rubella syndrome: A brief review of public health perspectives. Indian J Public Health. 2018;62(1):52-54. doi:10.4103/ijph.IJPH_275_16
- Yazigi A, De Pecoulas AE, Vauloup-Fellous C, Grangeot-Keros L, Ayoubi JM, Picone O. Fetal and neonatal abnormalities due to congenital rubella syndrome: a review of literature. J Matern-Fetal Neonatal Med Off J Eur Assoc Perinat Med Fed Asia Ocean Perinat Soc Int Soc Perinat Obstet. 2017;30(3):274-278. doi:10.3109/14767058.2016.1169526
- Armstrong NT. The ocular manifestations of congenital rubella syndrome. Insight Am Soc Ophthalmic Regist Nurses. 1992;17(1):14-16.
- Meyer BI, Liao A, Sanda GE, et al. Fundus imaging features of congenital rubella retinopathy. Graefes Arch Clin Exp Ophthalmol Albrecht Von Graefes Arch Klin Exp Ophthalmol. 2024;262(3):777-788. doi:10.1007/s00417-023-06284-x
- Tsang S, Sharma T. Rubella Retinopathy. In: Atlas of Inherited Retinal Diseases. Vol 1085. Advances in Experimental Medicine and Biology. Springer, Cham; 2018:215-217. https://doi.org/10.1007/978-3-319-95046-4_45
- Jain R, Daigavane S. Salt and Pepper Pigmentary Retinopathy in Congenital Rubella Syndrome: A Case Report. J Clin Diagn Res. Published online 2024. doi:10.7860/JCDR/2024/67103.19506
- Frank KE, Purnell EW. Subretinal neovascularization following rubella retinopathy. Am J Ophthalmol. 1978;86(4):462-466. doi:10.1016/0002-9394(78)90290-8
- Goldberg N, Chou J, Moore A, Tsang S. Autofluorescence Imaging in Rubella Retinopathy. Ocul Immunol Inflamm. 2009;17(6):400. doi:10.3109/09273940903118634
Faculty Approval by: Elizabeth Fairless, MD
Copyright Statement: Alina Husain, Elizabeth Fairless, ©2024. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
Suprachoroidal Hemorrhage as a Complication of Routine Cataract Surgery
Title: Suprachoroidal Hemorrhage as a Complication of Routine Cataract Surgery
Authors: Chris Wallace-Carrete, BS; Paul Israelsen, MD; Iqbal Ike K. Ahmed, MD; Jeff Pettey, MD
Date: 7/11/2024
Keywords/Main Subjects: Suprachoroidal hemorrhage (SCH), phacoemulsification, expulsive hemorrhage, cataract, cataract surgery, surgical complications
Diagnosis: Suprachoroidal Hemorrhage
Description of Case: An 85-year-old male with a history of age-related cataract in both eyes presented for cataract surgery in the right eye (OD). The patient had a history of cutaneous melanoma and basal cell carcinoma but was otherwise healthy. He was not taking any medications and had no known drug allergies. His baseline eye examination OD was notable for visual acuity (VA) of 20/70 with correction, intraocular pressure (IOP) of 8 mmHg, 2-3+ nuclear sclerotic cataract, and a manifest refraction with spherical equivalent of -3.75 diopters. Fundus examination OD demonstrated a cupless disc as well as an engorged vortex vein with pigmentary changes in the periphery. Preoperative examination demonstrated normal vital signs except for an elevated blood pressure at 152/81. On biometry OD, his axial length was 24.21 mm.
The patient was scheduled for routine cataract extraction with IOL insertion OD. The surgery progressed in the expected manner until the epinuclear removal stage when it was noted that subtle folds were developing in the posterior capsule (1. Epinuclear Removal_MORAN_07112024). The operating surgeon recognized that the posterior capsular folds were likely indicative of increased posterior pressure; therefore, viscoelastic was used to deepen the anterior chamber (2. Viscoelastic Insertion_MORAN_07112024). As viscoelastic was being injected into the anterior chamber, there was a large egress of viscoelastic back out through the primary wound, as well as mild iris prolapse. The eye was noted to be firm despite minimal viscoelastic remaining in the anterior chamber. The red reflex remained unchanged. When asked, the patient endorsed an aching pain in the right eye that began at roughly the same time that the posterior capsular folds appeared. His blood pressure was noted to be elevated at 153/62. The operating surgeon made the decision at this point to stop the surgery due to concern for suprachoroidal hemorrhage. The surgical wounds were quickly sealed by hydration (3. Wound Hydration_MORAN_07112024) and the remaining viscoelastic was removed from the eye (4. Viscoelastic Removal_MORAN_07112024). A fundus examination was performed with indirect ophthalmoscopy while the patient was still on the operating table, and a suprachoroidal hemorrhage in the temporal periphery was confirmed. The surgery was concluded, and the patient was left aphakic. The patient was then transferred to the post-anesthesia care unit.
During a same-day post-operative examination of the right eye, the patient was noted to have a VA of count fingers at four feet. His IOP was elevated at 41 mmHg and a dilated fundus examination (DFE) demonstrated a stable temporal choroidal hemorrhage. The patient was prescribed oral Diamox (acetazolamide) along with prednisolone and moxifloxacin eyedrops and was given instructions to return the following day for a repeat examination. On post-operative day (POD) 1, the patient’s VA was unchanged; however, his pressure had returned to his baseline of 8 mmHg. The suprachoroidal hemorrhage remained unchanged on DFE. Diamox was discontinued, and the patient was scheduled for follow-up in one week. On POD 4, the patient’s eye exam remained unchanged from POD 1. He received a B-scan ultrasound which confirmed a focal choroidal hemorrhage temporally OD. The patient’s SCH was monitored over a period of several weeks with plans for IOL insertion upon resolution of the hemorrhage.
A suprachoroidal hemorrhage (SCH) is a rare complication of intraocular surgery that develops when blood accumulates in the potential space that exists between the sclera and choroid. Intraoperative SCH is not a common occurrence of cataract surgery. There is an incidence of 0.03 – 0.13%, which has decreased over the last 25 years with the introduction of less invasive phacoemulsification techniques.1 Risk factors for the development of an intraoperative SCH include advanced age, uncontrolled hypertension, prolonged intraocular hypotony, increased surgical time, high myopia, nanophthalmos, and a history of SCH.2,3 Although the pathophysiology of SCH is not completely understood, it is hypothesized that low intraocular pressure during ocular surgery leads to an environment where the long and short posterior ciliary arteries and veins stretch and can eventually rupture, leading to rapid accumulation of blood in the suprachoroidal space.4 Major signs and symptoms of intraoperative SCH include shallowing of the anterior chamber, bulging of the posterior capsule, loss of the red reflex (5. SCH with Loss of Red Reflex_AHMED), sudden onset of ocular pain and/or headache, and expulsion of intraocular contents (6. Expulsive Hemorrhage_ISRAELSEN).2,4 Prompt intervention at the onset of SCH is vital to prevent progression and preserve vision. As soon as a SCH is suspected, the surgeon should immediately pressurize the eye to tamponade the ensuing hemorrhage.4,5 This is typically accomplished through prompt closure of all surgical wounds with either hydration or sutures. Additional perioperative management includes addressing systemic blood pressure with pharmacologic intervention as well as controlling IOP, allowing for normotonic or slightly hypertonic pressures.4,6
Management in the postoperative period includes a combination of pharmacotherapy and close monitoring with B-scan ultrasound and dilated fundus examination.4,7 A SCH is confirmed on B-Scan if there is hyperechoic material within a choroidal detachment. Often, a SCH will resolve over time with observation. Surgical management may be indicated in cases where the SCH is particularly severe (i.e. kissing choroidals), when the hemorrhage continues to progress despite initial management, the IOP remains uncontrollably elevated, or if the patient experiences intractable pain.8 Surgery to drain a choroidal hemorrhage is typically performed 7-14 days after the initial occurrence to allow for liquefaction of blood in the suprachoroidal space. Surgery is performed by creating a full thickness sclerotomy in the area of the greatest accumulation of suprachoroidal blood to allow for drainage. This is often followed by pars plana vitrectomy.
Prophylactic measures to mitigate the risk of SCH are focused on managing patient risk factors such as uncontrolled hypertension, atherosclerosis, and the use of anticoagulant or antiplatelet medications.4 The outcome of SCH varies depending on the severity of the hemorrhage and promptness of intervention.9 In some cases, such as phacoemulsification-related SCH, the visual prognosis is often excellent.10 Severe SCH often portends a poor prognosis with irreversible vision loss.11
To help minimize the risk of vision loss from SCH, all ophthalmic surgeons should attempt to mitigate any significant preoperative risk factors, be able to recognize signs of intraoperative SCH, and react promptly when a SCH is suspected to prevent progression.
Images or video:
- Epinuclear Removal: (1. Epinuclear Removal_MORAN_07112024)
- Cortical cleaving hydrodissection is used to remove the remaining portions of the lens cortex. At the end of phacoemulsification, subtle folds begin to develop in the posterior capsule with concurrent capsule bulging, indicating increased posterior segment pressure.
- Cortical cleaving hydrodissection is used to remove the remaining portions of the lens cortex. At the end of phacoemulsification, subtle folds begin to develop in the posterior capsule with concurrent capsule bulging, indicating increased posterior segment pressure.
- Viscoelastic Insertion: (2. Viscoelastic Insertion_MORAN_07112024)
- Viscoelastic is inserted into the lens capsule via cannula to deepen the anterior chamber. As viscoelastic is inserted into the eye, there is a large egress of viscoelastic back through the primary wound, further indicating increased posterior segment pressure. There is also a subtle iris prolapse toward the primary wound. The cannula is removed from the eye and used to check eye firmness. The eye is noted to be very firm and IOP is elevated at 40 mmHg. Due to the positive pressure in the eye and inadequate space to place the intraocular lens, no lens was inserted into the eye, and the decision was made to end the surgery.
- Viscoelastic is inserted into the lens capsule via cannula to deepen the anterior chamber. As viscoelastic is inserted into the eye, there is a large egress of viscoelastic back through the primary wound, further indicating increased posterior segment pressure. There is also a subtle iris prolapse toward the primary wound. The cannula is removed from the eye and used to check eye firmness. The eye is noted to be very firm and IOP is elevated at 40 mmHg. Due to the positive pressure in the eye and inadequate space to place the intraocular lens, no lens was inserted into the eye, and the decision was made to end the surgery.
- Wound Hydration: (3. Wound Hydration_MORAN_07112024)
- Pressurization of the eye is reestablished as the two surgical wounds are promptly closed via hydration.
- Pressurization of the eye is reestablished as the two surgical wounds are promptly closed via hydration.
- Viscoelastic Removal: (4. Viscoelastic Removal_MORAN_07112024)
- To prevent post-operative IOP spikes, the I/A handpiece was used to extract the viscoelastic. The corneal wounds were again sealed and subsequently checked with a weck-cel and were found to be watertight.
- To prevent post-operative IOP spikes, the I/A handpiece was used to extract the viscoelastic. The corneal wounds were again sealed and subsequently checked with a weck-cel and were found to be watertight.
- Example of Intraoperative Suprachoroidal Hemorrhage during Phacoemulsification (5. SCH with Loss of Red Reflex_AHMED)
- Complicated cataract surgery demonstrating a suprachoroidal hemorrhage. A classic yet not always present sign of SCH is loss of the red reflex, which can be seen during this procedure as the ensuing hemorrhage extends toward the equator. (Video courtesy of Iqbal Ike K. Ahmed M.D.)
- Complicated cataract surgery demonstrating a suprachoroidal hemorrhage. A classic yet not always present sign of SCH is loss of the red reflex, which can be seen during this procedure as the ensuing hemorrhage extends toward the equator. (Video courtesy of Iqbal Ike K. Ahmed M.D.)
- Example of Expulsive Hemorrhage (6. Expulsive Hemorrhage_ISRAELSEN)
- A severe outcome of a suprachoroidal hemorrhage is expulsion of intraocular contents through the surgical wounds. An example of an expulsive hemorrhage can be seen during this complex corneal transplant. (Video courtesy of Paul Israelsen, M.D.)
- A severe outcome of a suprachoroidal hemorrhage is expulsion of intraocular contents through the surgical wounds. An example of an expulsive hemorrhage can be seen during this complex corneal transplant. (Video courtesy of Paul Israelsen, M.D.)
Summary of the Case: An 85-year-old male with a history of age-related cataract in both eyes underwent routine cataract surgery in the right eye. He developed a suprachoroidal hemorrhage (SCH) during the intraoperative period shortly after the cataract was removed. Initial signs of the SCH included posterior capsule bulging, anterior chamber shallowing, mild iris prolapse, and right-sided ocular pain. Recognition of these signs and prompt closure of the surgical wounds prevented further progression of the SCH, which was confirmed with fundus examination before the patient left the operating room. B-scan ultrasound in the postoperative period allowed for characterization and monitoring of the hemorrhage. The patient’s risk factors included advanced age and elevated intraoperative blood pressure.
Suprachoroidal hemorrhage is a rare complication of intraocular surgery. Prompt recognition of the intraoperative signs of SCH and immediate intervention whenever a SCH is suspected are necessary to prevent irreversible vision loss. Key intraoperative signs of SCH include anterior chamber shallowing, loss of the red reflex, posterior capsule bulging, patient discomfort, and expulsion of intraocular contents. Immediate management of a SCH typically consists of pressurization of the eye by closure of all surgical wounds. B-scan ultrasound is important for diagnosis and monitoring in the postoperative period. In some instances, surgical management through drainage of suprachoroidal blood may be indicated. Prognosis varies based on the severity of the hemorrhage and the promptness of intervention.
Format: Video
References:
- Savastano A, Rizzo S, Savastano MC, et al. Choroidal Effusion and Suprachoroidal Hemorrhage during Phacoemulsification: Intraoperative Management to Prevent Expulsive Hemorrhage. European Journal of Ophthalmology. 2016;26(4):338-341. doi:10.5301/ejo.5000707
- Foo R, Tsai A, Lim L. Management of suprachoroidal hemorrhage. American Academy of Ophthalmology Web site.https://www.aao.org/eyenet/article/management-of-suprachoroidal-hemorrhage. Updated 2018.
- Wolter JR, Garfinkel RA. Ciliochoroidal effusion as precursor of suprachoroidal hemorrhage: A pathologic study. Ophthalmic Surg Lasers Imaging Retina. 1988;19(5):344-349.
- Chu, T. G., & Green, R. L. (1999). Suprachoroidal Hemorrhage. Survey of Ophthalmology, 43(6), 471–486. https://doi.org/10.1016/S0039-6257(99)00037-5
- Mantopoulos D, Hariprasad SM, Fine HF. Suprachoroidal hemorrhage: Risk factors and diagnostic and treatment options. Ophthalmic Surg Lasers Imaging Retina. 2019;50(11):670-674. doi: 10.3928/23258160-20191031-01 [doi].
- Koksaldi, S.; Utine, C.A.; Kayabasi, M. Management of Suprachoroidal Hemorrhage during Cataract Surgery: A Case Report. Beyoglu Eye 2022, 7, 66–70.
- Beatty, S.; Lotery, A.; Kent, D.; O’Driscoll, A.; Kilmartin, D.J.; Wallace, D.; Baglivo, E. Acute intraoperative suprachoroidal haemorrhage in ocular surgery. Eye 1998, 12, 815–820.
- Learned D, Eliott D. Management of delayed suprachoroidal hemorrhage after glaucoma surgery. Semin Ophthalmol. 2018;33(1):59-63. doi: 10.1080/08820538.2017.1353814 [doi].
- Wang L, Yang C, Yang C, et al. Clinical characteristics and visual outcome of non-traumatic suprachoroidal haemorrhage in taiwan. Acta Ophthalmol. 2008;86(8):908-912. doi: 10.1111/j.1755-3768.2008.01266.x.
- Gupta, A.; Ionides, A. Does acute suprachoroidal haemorrhage during phacoemulsification cataract surgery need surgical treatment? Graefes Arch. Exp. Ophthalmol. 2022, 260, 3395–3396.
- Qureshi, A.; Jalil, A.; Sousa, D.C.; Patton, N.; Dhawahir-Scala, F.; Charles, S.J.; Turner, G.; Ivanova, T. Outcomes of suprachoroidal haemorrhage drainage with and without vitrectomy: A 10-year study. Eye 2020, 35, 1879–1885.
Faculty Approval by: Dr. Jeff Pettey
Copyright statement: Chris Wallace-Carrete, Paul Israelsen, Iqbal Ike K. Ahmed, Jeff Pettey, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Lens-Induced Glaucoma: Overview and Case Report
Title: Lens-Induced Glaucoma: Overview and Case Report
Authors: Bryce Baugh, MSIV, University of Utah Spencer Fox Eccles School of Medicine, Austin Nakatsuka, MD
Date: 08/27/2024
Keywords/Main Subjects: lens-induced glaucoma, phacomorphic glaucoma, phacolytic glaucoma, phacoantigenic glaucoma, lens-particle glaucoma
Diagnosis: Phacomorphic Glaucoma
Description of Case:
A 64-year-old male presented with five days of acute vision loss and light sensitivity in the right eye. The patient reported that when he awoke in the morning five days prior, he was unable to see out of his right eye. The patient denied any pain or irritation but noted an increased pressure sensation in his right eye. He denied any redness or discharge. Since initial onset, his vision had slightly improved. His ocular history was notable for retinal detachment 20 years prior with cryotherapy. Ocular history was also notable for LASIK more than 20 years prior complicated by increased IOP due to a steroid response following surgery. Other past medical history was non-contributory. The patient’s visual acuity at initial presentation was 20/80 in the right eye, 20/30 in the left eye. Applanation revealed a pressure of 40 in the right eye, 10 in the left eye. Gonioscopy showed A20b in the right eye and C35f 2+ptm in the left eye circumferentially. Visual field defects in right superonasal and superotemporal fields were noted. Slit lamp exam of the right eye showed trace conjunctival injection, hazy cornea, very narrow anterior chamber, and 2+ nuclear sclerosis and 2+ cortical cataract. The fundus exam showed tilted disc, with tilted cup and small optic nerve, and significant nasal peripapillary atrophy. The left eye showed 1+ nuclear sclerosis and was otherwise normal.
The decision was made to proceed with laser peripheral iridotomy of the right eye for treatment of acute angle closure glaucoma, likely phacomorphic in nature. Following iridotomy, the patient was instructed to continue Cosopt and Brimonidine twice daily in the right eye, as well as acetazolamide 500mg twice daily. The following day, the pressure in the right eye was 7 and visual acuity was 20/70. The anterior chamber still appeared narrow, but now appeared more even centrally and peripherally. The patient was instructed to continue all medications. At follow up the following week, the patient’s vision had improved to 20/30 and pressure in the right eye was 11. With pressures now under control but persistently shallow-appearing anterior chamber and mature cataract, the patient was scheduled for phacoemulsification and IOL placement with goniotomy and anterior vitrectomy of the right eye. This took place approximately 1 month after the patient’s initial presentation.
Intraoperatively, it was noted that the patient had a very shallow anterior chamber, with little space to even make corneal incisions. During phacoemulsification, it was noted that the lens capsule had significant global zonular laxity necessitating the placement of a capsular tension ring before lens placement. A nasal 3-4 clock hour goniotomy with the Kahook Dual Blade was done after the phacoemulsification and lens placement were completed. Please see attached surgical video with narration for more details.
At the patients first postoperative visit 1 week following surgery, pinhole visual acuity was 20/60 in the right eye, and pressure was 15. The anterior chamber appeared deep, with 1+ cell. The patient was continued on brimonidine BID OD, and prednisolone BID OD for 2 weeks. The patient appears clinically improved and will continue to be monitored at subsequent follow up appointments.
Overview:
Lens-induced glaucoma is form of secondary glaucoma where the ocular lens is directly involved in the mechanism contributing to increased intra-ocular pressure (IOP). It is one of the most common types of secondary glaucoma presenting with a senile cataract. There are thought to be four primary processes through which this can occur–one involving aqueous obstruction, and the others being lens-protein associated. The different etiologies include phacomorphic, phacolytic, lens-particle associated, and phacoantigenic. The pathogenesis, clinical features, and management are outlined below.
Pathogenesis:
Phacomorphic glaucoma is the most common type of lens-induced glaucoma and involves a sudden increase in the volume of the cataract lens. The intumescent lens can lead to pupillary block and disruption of aqueous humor flow from the posterior to anterior chamber. As pressure builds in the posterior chamber, the iris is pushed forward, obstructing outflow through the trabecular meshwork and causing acute angle closure.1
Phacolytic glaucoma is thought to involve obstruction of the trabecular meshwork by macrophages and typically occurs in the setting of a mature senile cataract. High molecular weight proteins leak into the anterior chamber from microdefects in the lens capsule. The macrophages attempting to phagocytose these proteins block the trabecular meshwork, resulting in an open-angle glaucoma.2
Lens-particle glaucoma typically presents following trauma, cataract surgery, or YAG capsulotomy. As lens fragments are left behind, they migrate and deposit in the trabecular meshwork, resulting in obstruction of outflow and an open-angle glaucoma.
Phacoantigenic glaucoma is a type III hypersensitivity reaction and typically presents 1-14 days following cataract surgery.3 It is normally associated with a complicated cataract surgery involving the mixture of lens and vitreous.4 As lens particles are released, there is a sensitization period where immune complexes are formed. The reaction promotes inflammatory cells which subsequently block the trabecular meshwork causing open-angle glaucoma.
Clinical Features:
| Symptoms | Signs | |
| Phacomorphic Glaucoma |
· Severe eye pain
· Blurry vision · Eye redness · Headache · Nausea/vomiting
|
· Conjunctival injection
· Corneal edema · Mid-dilated pupil · Intumescent lens pushing iris forward · Decreased AC depth
|
| Phacolytic Glaucoma |
· Severe eye pain
· Blurry vision · Eye redness
|
· Corneal edema
· Fresh karatitic precipitates on posterior surface of cornea · AC cell and flare · Hyper-refringent particles composed of calcium oxalate or cholesterol crystals5 · Anterior peripheral/posterior synechiae · Hypermature cataract
|
| Lens-Particle Glaucoma |
· Severe eye pain following trauma or surgery
· Blurry vision · Eye redness |
· Conjunctival injection
· Corneal edema · Anterior peripheral/posterior synechiae · Lens proteins deposited on posterior surface of cornea · Lens particles in AC · Rupture of the lens capsule with a cataractous lens may be present
|
| Phacoantigenic Glaucoma |
· Severe eye pain
· Blurry vision · Eye redness
|
· Eyelid edema
· Conjunctival injection · Corneal edema · Fresh karatitic precipitates on posterior surface of cornea · AC cell and flare · Anterior peripheral/posterior synechiae · Ruptured lens capsule vs. anterior capsulorrhexis with residual lens matter
|
Diagnostic tools:
- Applanation tonometry
- Gonioscopy
- B-scan if view of posterior segment limited
Treatment:
Phacomorphic Glaucoma:
- Lower IOP with aqueous suppressants or hyperosmotic agents
- Consider laser iridotomy
- Cataract surgery once IOP lowers and inflammation resides
Phacolytic Glaucoma:
- Topical steroids
- Topical aqueous suppressants to lower IOP
- Cataract surgery is definitive treatment
Lens-particle Glaucoma:
- If there is minimal cortical material: topical steroids, cycloplegics, topical aqueous suppressants
- If significant inflammation and IOP can’t be lowered quickly: early removal of residual lens cortex via irrigation or vitrectomy
Phacoantigenic Glaucoma:
- Topical steroids
- Topical aqueous suppressants to lower IOP
- Possible surgery to remove remanent lens material
Images or video:
- Images: Visual field of the right eye showing significant defect, most severe in the superonasal and superotemporal fields.


- Video: https://www.youtube.com/watch?app=desktop&v=j-NwR04ZJUI
- Narrated surgical video of cataract extraction and goniotomy in a patient with phacomorphic glaucoma.
Summary of the Case: Lens-induced glaucoma is form of secondary glaucoma where the ocular lens is directly involved in the mechanism contributing to increased intra-ocular pressure (IOP). The most common etiology is phacomorphic and other etiologies include phacolytic, lens-particle associated, and phacoantigenic. The presentation varies between etiologies, but largely presents with eye pain, blurry vision, eye inflammation, increased IOP, mature cataract, or following cataract surgery. Treatment focuses on timely reduction of IOP, minimizing inflammation, and eventual cataract surgery.
References:
- Ellant JP, Obstbaum SA. Lens-induced glaucoma. Doc Ophthalmol. 1992;81(3):317-338. doi:10.1007/BF00161770
- Epstein DL, Jedziniak JA, Grant WM. Obstruction of aqueous outflow by lens particles and by heavy-molecular-weight soluble lens proteins. Invest Ophthalmol Vis Sci. 1978;17(3):272-277.
- Perlman EM, Albert DM. Clinically unsuspected phacoanaphylaxis after ocular trauma. Arch Ophthalmol. 1977;95(2):244-246. doi:10.1001/archopht.1977.04450020045008
- Conner IP et al. Lens-induced glaucoma. In: Kahook M et al, eds. Chandler and Grant’s Glaucoma, 5th ed. Thorofare, N.J.; Slack; 2013:441-447
- Peracha-Riyaz MH, Peracha ZH, Spaulding J, et al. First Described Case of Anterior and Posterior Segment Crystals in Phacolytic Glaucoma. J Glaucoma. 2017;26(5):e171-e173. doi:10.1097/IJG.0000000000000642
Faculty Approval by: Austin Nakatsuka
Copyright statement: Bryce Baugh, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Giant Cell Arteritis
Home / Basic Ophthalmology Review / Retina
Title: Giant Cell Arteritis
Author: Michael Jensen, MSIV
Keywords/Main Subjects: Giant Cell Arteritis, AAION
Diagnosis: Giant Cell Arteritis
Description of Case: An 80-year-old male recently diagnosed with polymyalgia rheumatica presented to his primary ophthalmologist with complaints of 1 month of intermittent diplopia, greying of vision, severe bitemporal headache, scalp tenderness, malaise, and night sweats but denied jaw claudication. Eye exam was at baseline upon initial presentation with BCVA of 20/40 OD and 20/50 OS, no diplopia, and normal fundus exam. History and exam were most consistent a diagnosis of giant cell arteritis and the patient was prescribed 80mg oral prednisone. ESR, CRP, CBC were ordered and the patient was referred to an oculoplastic surgeon for a temporal artery biopsy the next morning. Overnight, patient experienced a sudden decrease in vision after taking his prednisone. On exam BCVA is now CF in both eyes and labs show elevation of ESR, CRP, and platelets. Retinal exam was significant for 1+ edema and 4+ pallor of the optic nerve. The patient was admitted to the hospital and started on 1000mg/day IV steroid treatment as well as adjuvant therapy with heparin, pentoxifylline, and brimonidine. Despite three days of maximal therapy, the patients vision declined to NLP OU.
Giant cell arteritis is a large-medium vessel vasculitis strongly associated with polymyalgia rheumatica. It primarily effects adults age 70 years and above and often presents with a temporal headache, scalp tenderness, jaw claudication, and vision changes. Other symptoms include throat, ear, or tongue pain, constitutional symptoms including fever, malaise, night sweats, fatigue, and proximal muscle soreness and weakness. Work up includes sensitive markers of inflammation like ESR, CRP, and platelets. Temporal artery biopsy is the gold standard for diagnosis but temporal artery ultrasound can also be performed by an experienced practitioner. Treatment includes immediate administration of steroids once the diagnosis is suspected, without waiting for biopsy results. Neuro-Ophthalmology and Rheumatology should be consulted in the in-patient setting. There is no strong evidence in favor of IV or oral steroids but common practice to treat with 1000 mg IV steroids for three days if there are accompanying vision changes. Regardless of route, early treatment has the largest impact on vision outcomes and delays in treatment can lead to permanent vision loss, like the patient described above. Adjuvant therapies like heparin are based on limited evidence and their use should be decided on a case-by-case basis as part of a multi-disciplinary team.
Summary of the Case: Classic presentation of giant cell arteritis is and older adult with history of PMR, temporal headache, vision changes, claudication within the distribution of the carotid arteries, and constitutional symptoms. Standard workup includes inflammatory markers and temporal artery biopsy. Steroids should be started immediately if there is a strong clinical suspicion. If exam is positive for visual changes, the patient should be admitted for IV steroids along with emergent neuro-ophthalmology and rheumatology consultation.
Images:

References:
- Salvarani, C., Pipitone, N., Versari, A., & Hunder, G. G. (2012). Clinical features of polymyalgia rheumatica and giant cell arteritis. Nature Reviews.Rheumatology, 8(9), 509-521. https://doi.org/10.1038/nrrheum.2012.97
- Gonzalez-Gay, M. A. , Barros, S. , Lopez-Diaz, M. J. , Garcia-Porrua, C. , Sanchez-Andrade, A. & Llorca, J. (2005). Giant Cell Arteritis. Medicine, 84 (5), 269-276. doi: 10.1097/01.md.0000180042.42156.d1.
- Hayreh SS, Zimmerman B. Management of giant cell arteritis. Our 27-year clinical study: new light on old controversies. Ophthalmologica. 2003 Jul-Aug;217(4):239-59. doi: 10.1159/000070631. PMID: 12792130
- Buono LM, Foroozan R, de Virgiliis M, Savino PJ. Heparin therapy in giant cell arteritis. Br J Ophthalmol. 2004 Feb;88(2):298-301. doi: 10.1136/bjo.2003.021592. PMID: 14736795; PMCID: PMC1772017
Faculty Approval by: Griffin Jardine, MD
Copyright statement: Copyright Michael Jensen, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Polypoidal Choroidal Vasculopathy
Home / Retina and Vitreous / Age-Related Macular Degeneration and other Causes of Choroidal Neovascularizaton
Title: Polypoidal Choroidal Vasculopathy
Authors: Marlow Schulz, BS; Brian Solinsky, MD; Paul Bernstein, MD
Date: June 23, 2024
Keywords/Main Subjects: Polypoidal Choroidal Vasculopathy, PCV
Diagnosis: Polypoidal Choroidal Vasculopathy (PCV)
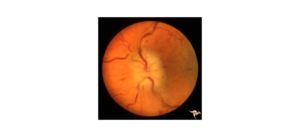
Description of Case: This patient is a 64-year-old male of Asian heritage with an ocular history of high myopia and no significant past medical history presenting with one year of gradually worsening vision in his left eye. He reported that around three weeks ago, the vision in his left eye acutely became even more blurry, prompting him to present for evaluation. He denied flashes of light or floaters in his vision, denies any vision changes in the right eye, and denies recent head or eye trauma. His best corrected visual acuity on presentation was 20/15 OD and 20/80 OS. Tonometry, pupils, visual fields, and extraocular movements were all normal. Anterior chamber exam was notable for trace nuclear sclerosis of the lens, otherwise unremarkable. Color fundus photography demonstrated subretinal pigment epithelium and diffuse macular exudates (A). Optical coherence tomography (OCT) demonstrated significant sub-retinal fluid (SRF) with sub-foveal fluid (B), and a large fibrovascular pigment epithelial detachment in the temporal macula. Fluorescein angiogram (FA) showed a large area of hyperfluorescense in the temporal macula with leakage (C) and Indocyanine Green Angiography (ICG) demonstrated a polyp in the choroidal vasculature (D). The patient was subsequently treated with intravitreal Avastin and photodynamic therapy. Post-treatment OCT showed a significant reduction in SRF (D), FA demonstrated resolution of leakage (E).
Polypoidal choroidal vasculopathy (PCV) is a macular disease characterized by an abnormality in the choroidal blood vessels. This condition is considered a subtype of age-related macular degeneration (AMD), though it has distinct clinical features. Of note, it is more commonly diagnosed in patients of Asian or African heritage, with 23-62% of those with neovascular AMD also carrying this diagnosis, compared to 8-13% of Caucasian patients.
The hallmark of PCV is the presence of polyp-like, dilated blood vessels in the choroid, typically located at the border of branching vascular networks. PCV can cause fluid accumulation or bleeding beneath the retina, leading to serous or hemorrhagic detachments respectively. This can lead to blurred or distorted vision, scotomas in the central vision, as well as a sudden decreased in vision related to new fluid accumulation. Treatment includes a combination of anti-VEGF therapy to reduce fluid and stabilize abnormal blood vessels, photodynamic therapy to selectively target abnormal blood vessels, as well as laser photocoagulation in some cases. PCV requires ongoing monitoring and treatment to manage symptoms and preserve vision, as it can be a chronic and recurring condition.
Images:

A. Color fundus photography at presentation
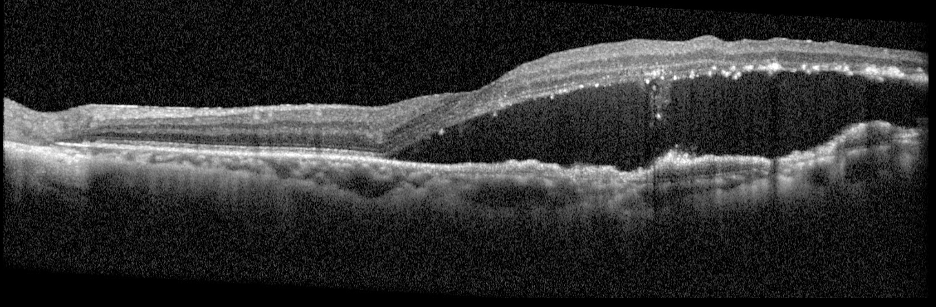
B. OCT images at presentation
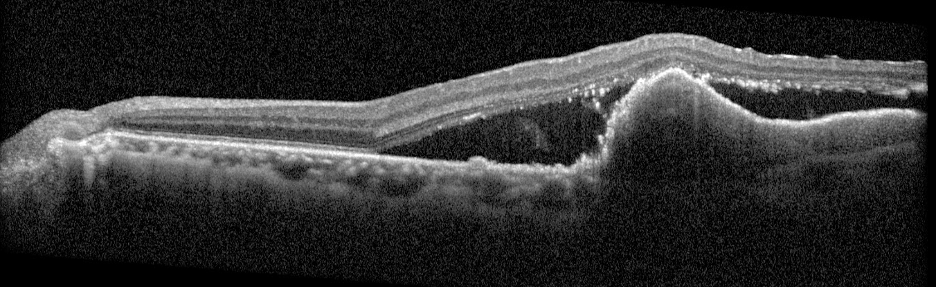
B. OCT images at presentation
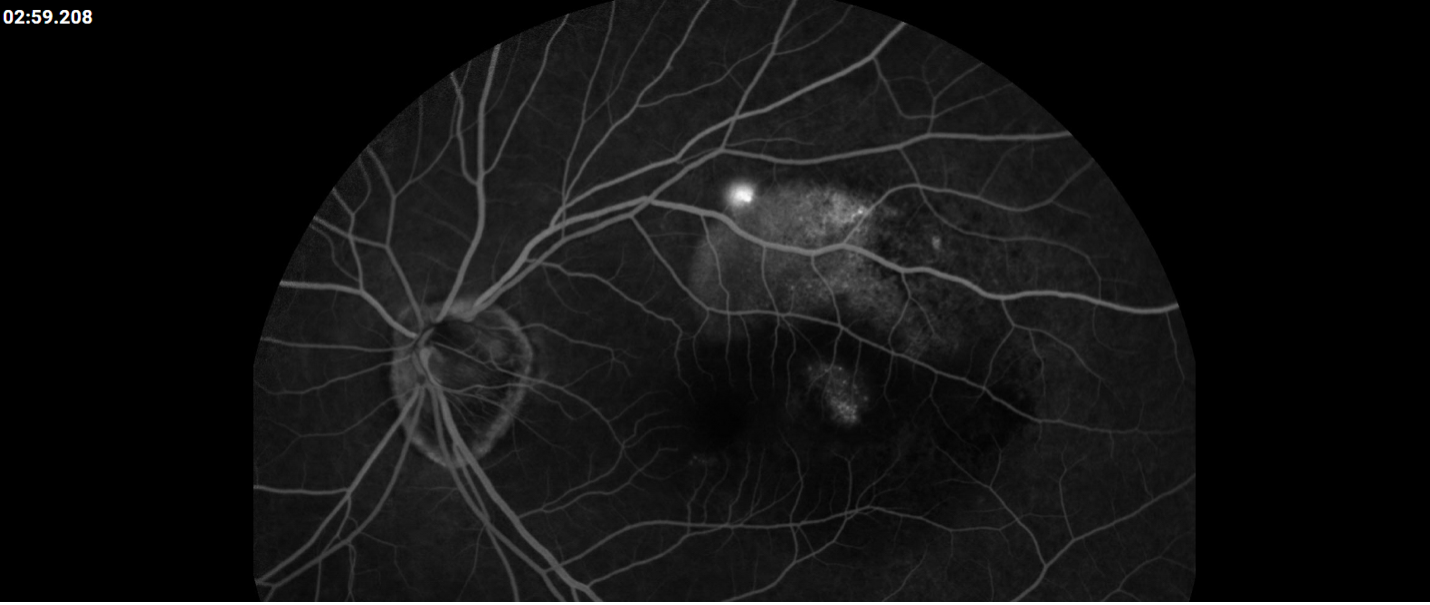
C. FA at presentation

C. FA at presentation
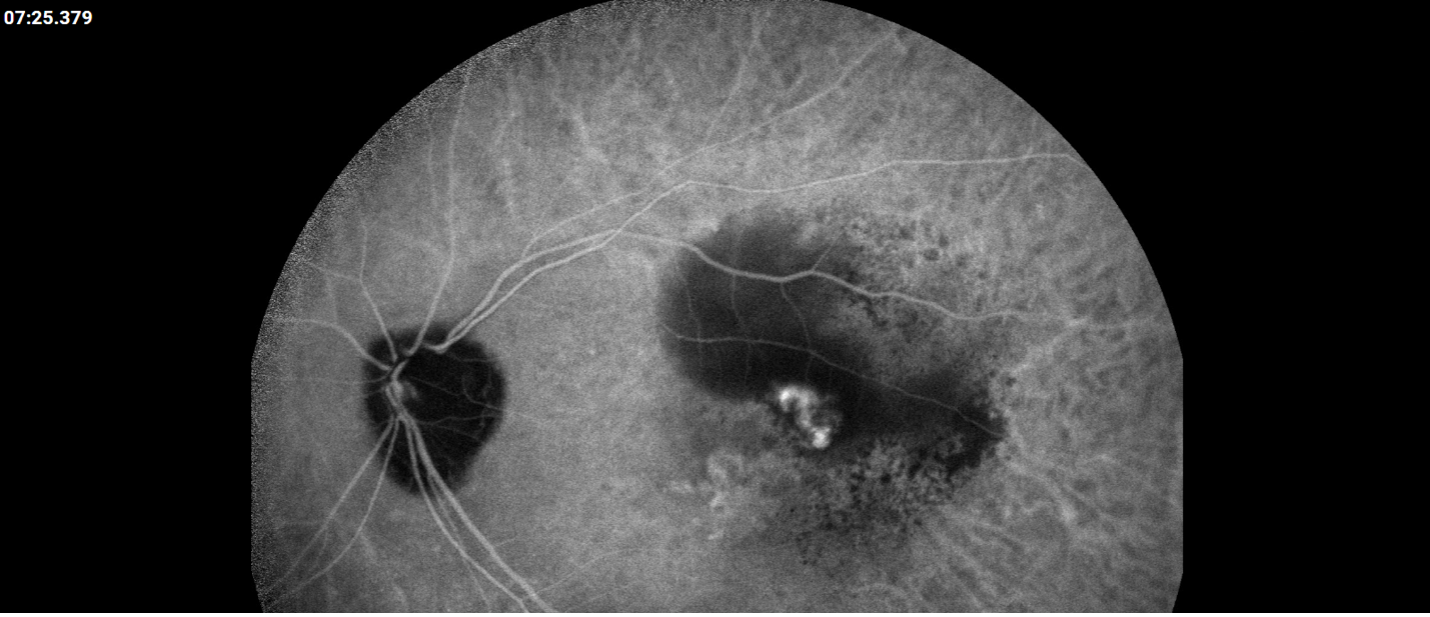
D. ICG at presentation
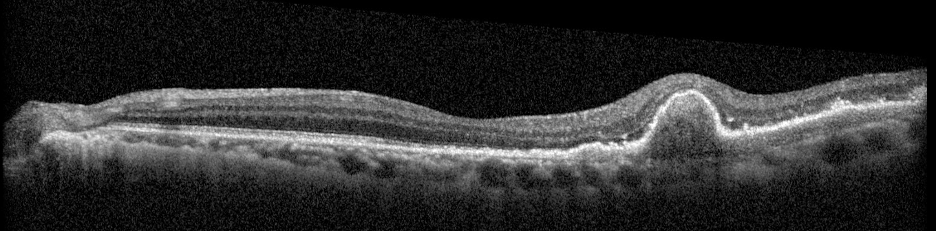
E. Post-treatment OCT
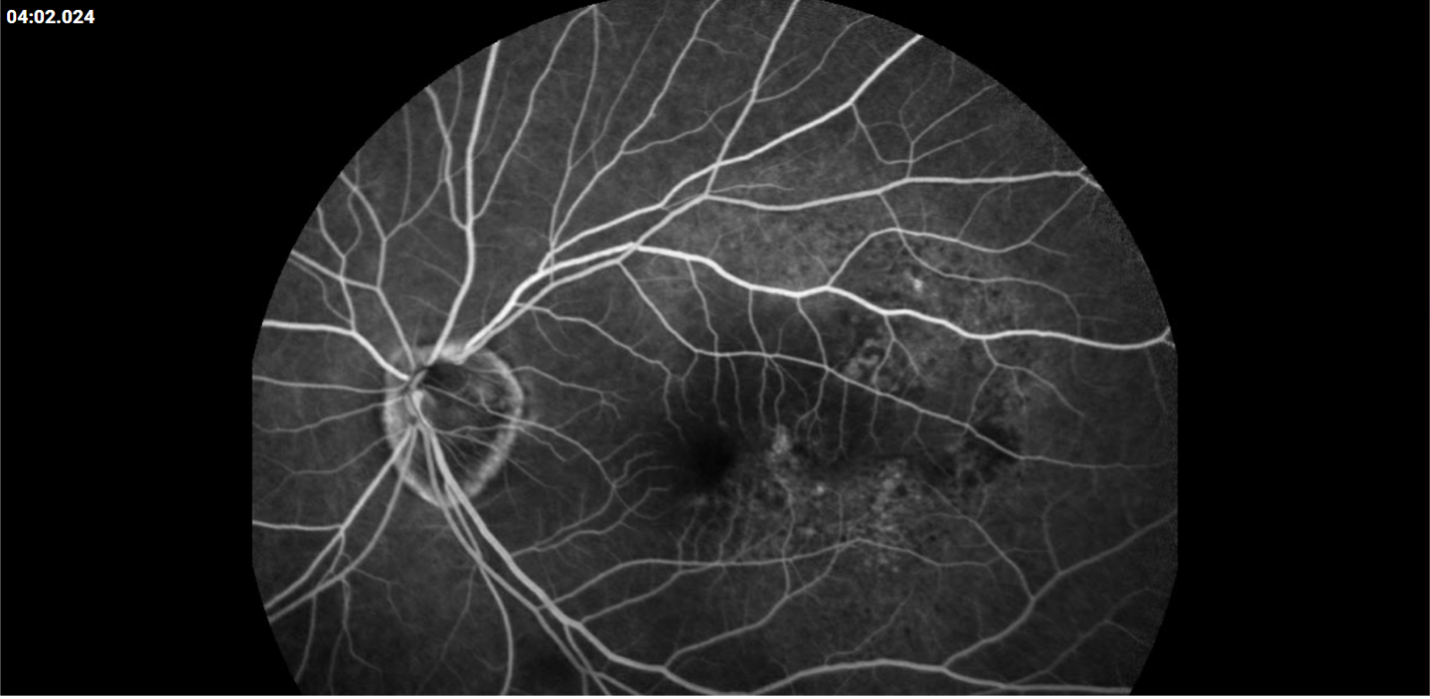
F. Post-treatment FA
Summary of the Case: Polypoidal choroidal vasculopathy (PCV) is a subtype of age-related macular degeneration (AMD) more prevalent among patients of Asian or African heritage, accounting for 23-62% of neovascular AMD cases in these populations compared to 8-13% in Caucasian patients. PCV is characterized by the presence of polyp-like, dilated blood vessels in the choroid, which can lead to fluid accumulation or bleeding beneath the retina. This condition often manifests as blurred or distorted vision, scotomas, and sudden vision decrease due to new fluid accumulation. Treatment for PCV typically involves a combination of anti-VEGF therapy to reduce fluid and stabilize abnormal blood vessels, photodynamic therapy to selectively target these vessels, and sometimes laser photocoagulation. Due to its chronic and recurring nature, PCV requires ongoing monitoring and treatment to manage symptoms and preserve vision.
References:
- Chawla H, Blair K, Vohra V. Polypoidal Choroidal Vasculopathy. [Updated 2023 Mar 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK567780/
- Wong RL, Lai TY. Polypoidal choroidal vasculopathy: an update on therapeutic approaches. J Ophthalmic Vis Res. 2013 Oct;8(4):359-71. PMID: 24653824; PMCID: PMC3957043.
- Wong CW, Wong TY, Cheung CM. Polypoidal Choroidal Vasculopathy in Asians. J Clin Med. 2015 Apr 24;4(5):782-821. doi: 10.3390/jcm4050782. PMID: 26239448; PMCID: PMC4470199.
Faculty Approval by: Paul Bernstein, MD
Copyright statement: Copyright Marlow Schulz; Brian Solinsky, MD; Paul Bernstein, MD, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Combined Retinal Vein and Artery Occlusion: A Case Series
Home / Retina and Vitreous / Other Retinal Vascular Disease
Title: Combined Retinal Vein and Artery Occlusion: A Case Series
Authors: Hansen Dang, BS; George Sanchez, MD; Brian Solinsky, MD; Akbar Shakoor, MD
Date: 8/12/2024
Keywords/Main Subjects: Retinal occlusion, cilioretinal artery, central retinal vein occlusion
Diagnosis: Combined retinal vein and artery occlusion
Description of Case:
Introduction
Central retinal vein occlusion (CRVO) is the second most common retinal vascular disorder that primarily affects older populations. In rare instances, CRVO can be accompanied by a retinal arterial occlusion leading to a combined retinal vein and arterial occlusion. This is a case series discussing two patients with this entity.
Case 1
A 28-year-old otherwise healthy female presented with sudden vision loss following a headache that occurred four days prior. She reported loss of central vision with intact peripheral vision of the left eye. Notably, she has a history of migraines associated with blurry vision changes lasting half the day. On exam, visual acuity of the left eye was counting fingers at face distance. Fundus exam of the left eye was remarkable for macular edema, retinal whitening with a cherry red spot, and marked dilation of venules with tortuosity; this was corroborated on fundus imaging (Figure 1). Humphrey visual field demonstrated a central scotoma. Fluorescein angiography was performed, and a diagnosis of combined CRVO and central retinal arterial occlusion (CRAO) of the left eye was made. Additionally, findings on ocular coherence tomography were consistent with a CRAO (Figure 2). She was evaluated by the neurology service. A complete stroke and coagulopathy workup, including computed tomography angiography head/neck, echocardiogram, and all pertinent markers of hypercoagulability, was completed. The workup was unremarkable other than a slightly elevated Factor VIII activity at 234% (normal range: 56-191%). She was discharged home on 81mg aspirin daily. Visual acuity was stable on follow-up.
Case 2
A 30-year-old female with hyperlipidemia presents to the emergency department with intermittent eye pain exacerbated with eye movements and worsening blurry vision in the left eye that started four days ago. She had two similar episodes in the past two weeks, but these episodes self-resolved. On exam, visual acuity in the left eye was 20/20 -1. Additional findings were remarkable for a relative afferent pupillary defect and mild red desaturation. Fundus exam was showed for grade VI optic disc edema, retinal whitening of the nasal macula along with the distribution of the cilioretinal artery, and diffuse venous tortuosity. These findings were also seen on fundus imaging (Figure 3). Virtual visual field showed inferior altitudinal and superior arcuate defects. Fluorescein angiography revealed the diagnosis of CRVO and cilioretinal artery occlusion (CLRAO) (Figure 4). OCT macula findings correlated with the CLRAO (Figure 5). Optic neuritis was originally suspected, but imaging did not reveal optic nerve enhancement. She was evaluated by the neurology service. A complete stroke and coagulopathy workup including computed tomography angiography head/neck, echocardiogram, and all pertinent markers of hypercoagulability, was completed. This was unremarkable, aside from an elevated antinuclear antibody level. The patient was discharged home on 81 mg aspirin daily with outpatient follow-up planned.
Discussion
The two patients presented were both cases of CRVO with retinal artery occlusion, albeit involving different arteries. Both patients were female and younger than the typical age for isolated CRVO patients, which is consistent with prior studies.(1, 2) Additionally, both were relatively healthy, each having only one identifiable clinical risk factor for retinal vein occlusion: migraines in case patient 1 and hyperlipidemia in case patient 2.(3) Coagulopathy panels were performed to rule out known risk factors for combined occlusion in this age group and were mostly unremarkable. Case patient 1 did, however, have elevated factor VIII activity, which has been shown to be a significant risk factor for venous thromboembolism, including CRVO.(4-6) There has been a documented case of a patient with combined CRVO and CRAO with elevated fibrinogen and factor VIII to 560%.(7) Notably, case patient 2 had an unremarkable workup that did not reveal underlying clotting tendency.
Despite their similarities, the two had drastically different visual outcomes, likely due to different arterial involvement. The mechanism of combined venous and arterial occlusions remains unclear, but a common explanation involves a reduction in retinal perfusion pressure.(1) The retina’s venous drainage is handled solely by the central retinal vein whereas the arterial supply is more variable. In the presence of a cilioretinal artery, the retina has two independent arterial sources: the central retinal artery arising from the ophthalmic artery and the cilioretinal artery arising from the posterior ciliary artery. If arterial pressures are greater than venous pressures, blood will flow appropriately, and the retina remains perfused. In the setting of CRVO, retinal venous pressure quickly rises. Therefore, to maintain perfusion, the arterial pressures must rise as well. The central retinal artery has no issues doing so due to its autoregulatory system. In response to decreased ocular perfusion pressure, local vascular dilation occurs in response to local oxygen and carbon dioxide tensions as well as pH level thereby leading to decreased vascular resistance and restoring adequate perfusion of the retina. This autoregulatory system is found in the central retinal artery. However, the cilioretinal artery does not have this luxury and thus is more prone to hypoperfusion. This was likely what happened in case patient 2. She demonstrated a cilioretinal artery occlusion without foveal involvement, resulting in better a visual outcome. For case patient 1, the CRVO likely caused a marked increase in venous pressure that overcame the central retinal artery’s autoregulatory capabilities, causing poor perfusion and larger retinal ischemia owed to the larger distribution of the central retinal artery. Therefore, the case patient had a worse visual outcome and prognosis. This patient, unlike case patient 2, did not have a cilioretinal artery.
Images:
Figure 1. Fundus photo of the left eye of case patient 1 showing retinal whitening with a cherry red macula and dilated tortuous vessels.
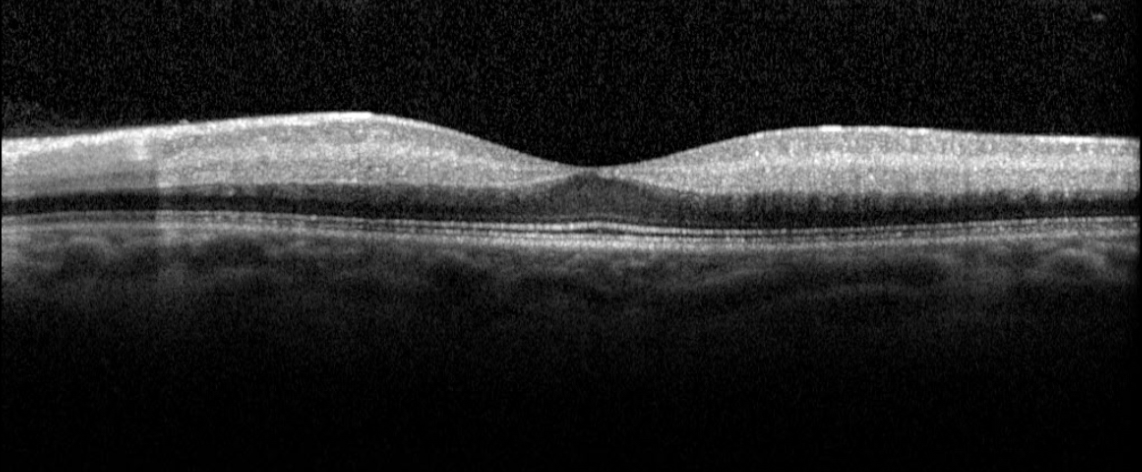
Figure 2. Optical coherence tomography macula of the left eye of case patient 1 showing inner retinal thickening, loss of architecture and hyperreflectivity consistent with central retinal artery occlusion.
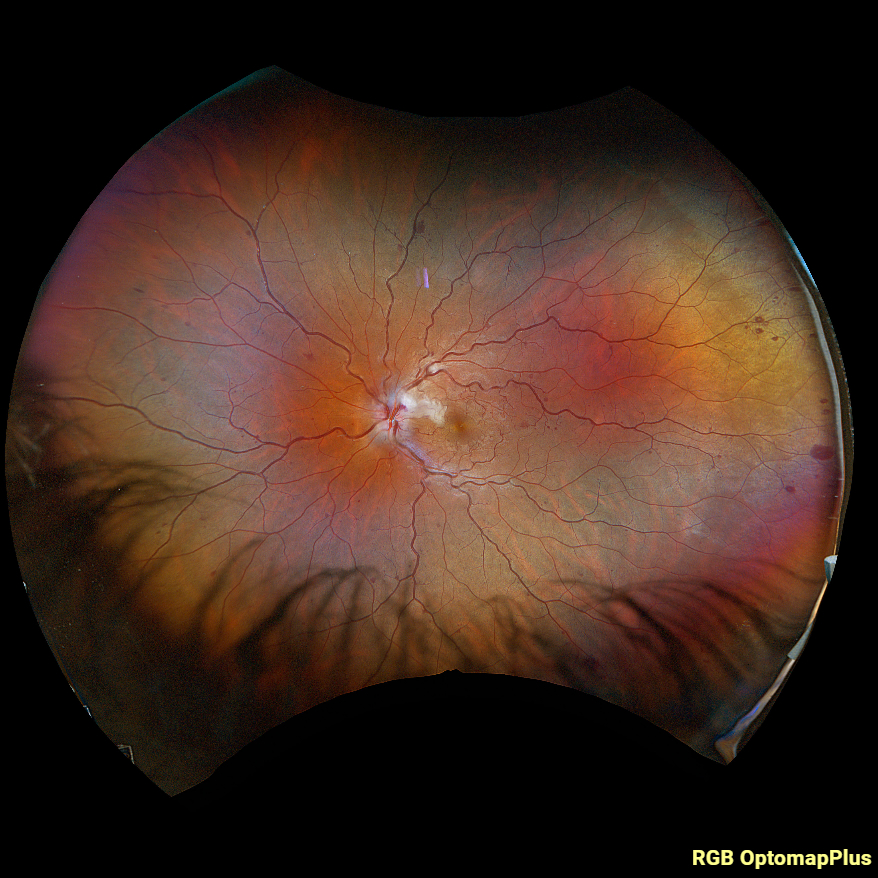
Figure 3. Fundus photo of the left eye of case patient 2 demonstrating retinal whitening along distribution of cilioretinal artery distribution.
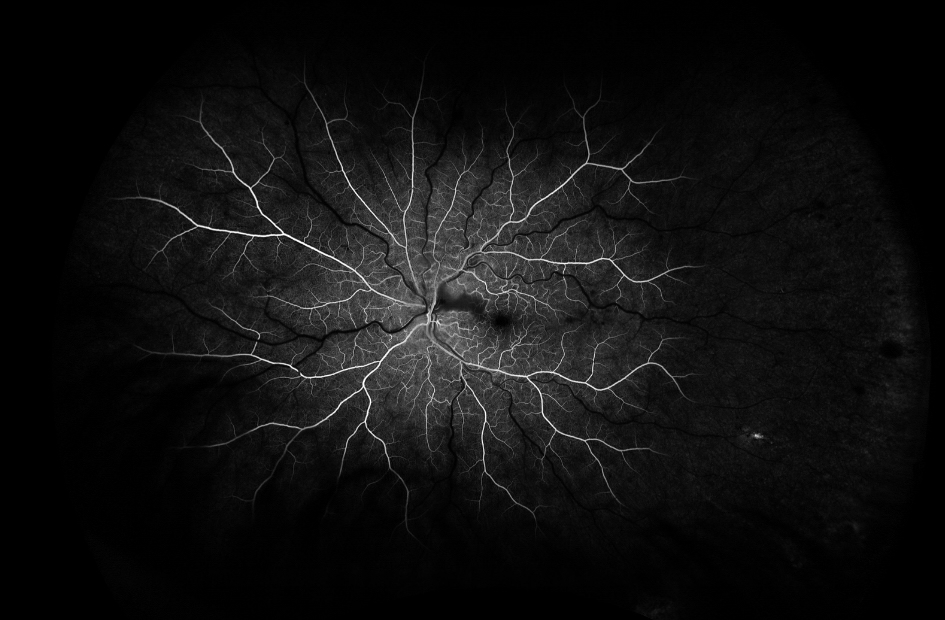
Figure 4. Fluorescein angiography of the left eye of case patient 2 showing no filling in the distribution of the cilioretinal artery.
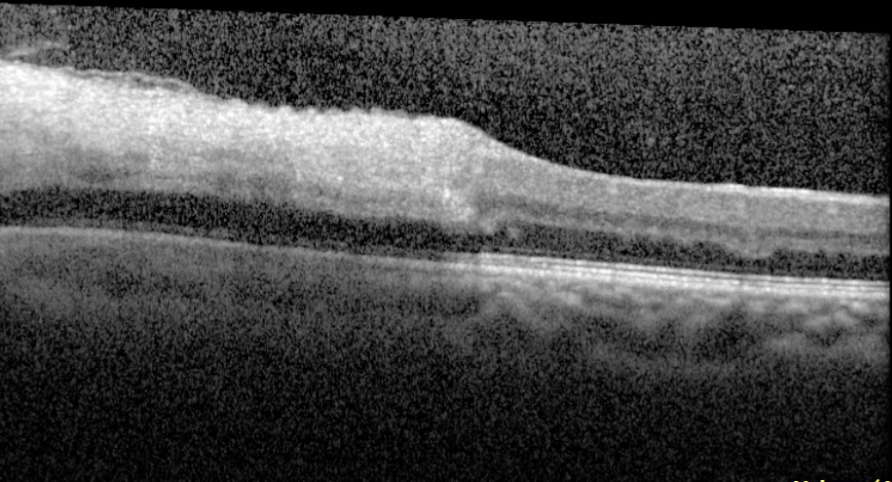
Figure 5. Optical coherence tomography macula of the left eye of case patient 2 showing inner retinal thickening and hyperreflectivity in the distribution of the cilioretinal artery.
Summary of the Case: Combined retinal vein and arterial occlusion is a rare condition with variable presentations and outcomes, depending on the specific artery and extent of involvement.
References:
- Hayreh SS, Fraterrigo L, Jonas J. Central retinal vein occlusion associated with cilioretinal artery occlusion. Retina 2008;28:581-594.
- Raval V, Nayak S, Saldanha M, Jalali S, Pappuru RR, Narayanan R, Das T. Combined retinal vascular occlusion: Demography, clinical features, visual outcome, systemic co-morbidities, and literature review. Indian J Ophthalmol 2020;68:2136-2142.
- Tilleul J, Glacet-Bernard A, Coscas G, Soubrane G, Souied EH. [Underlying conditions associated with the occurrence of retinal vein occlusion]. J Fr Ophtalmol 2011;34:318-324.
- Glueck CJ, Hutchins RK, Jurantee J, Khan Z, Wang P. Thrombophilia and retinal vascular occlusion. Clin Ophthalmol 2012;6:1377-1384.
- Kraaijenhagen RA, in’t Anker PS, Koopman MM, Reitsma PH, Prins MH, van den Ende A, Buller HR. High plasma concentration of factor VIIIc is a major risk factor for venous thromboembolism. Thromb Haemost 2000;83:5-9.
- Kyrle PA, Minar E, Hirschl M, Bialonczyk C, Stain M, Schneider B, Weltermann A, et al. High plasma levels of factor VIII and the risk of recurrent venous thromboembolism. N Engl J Med 2000;343:457-462.
- Chang IB, Lee JH, Kim HW. Combined Central Retinal Vein And Artery Occlusion In A Patient With Elevated Level Of Factor VIII: A Case Report. Int Med Case Rep J 2019;12:309-312.
Faculty Approval by: Dr. Akbar Shakoor, MD
Copyright statement: Copyright Hansen Dang ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Vogt-Koyanagi-Harada (VKH) Disease Case Report
Home / Intraocular Inflammation and Uveitis / Noninfections (Autoimmune) Uveitis
Title: Vogt-Koyanagi-Harada (VKH) Disease Case Report
Author: Taylor Johnson, MSIV, University of Utah Spencer Fox Eccles School of Medicine
Date: 8/10/2024
Keywords/Main Subjects: Vogt-Koyanagi-Harada Disease, uveitis
Diagnosis: Vogt-Koyanagi-Harada (VKH) Disease
Images:
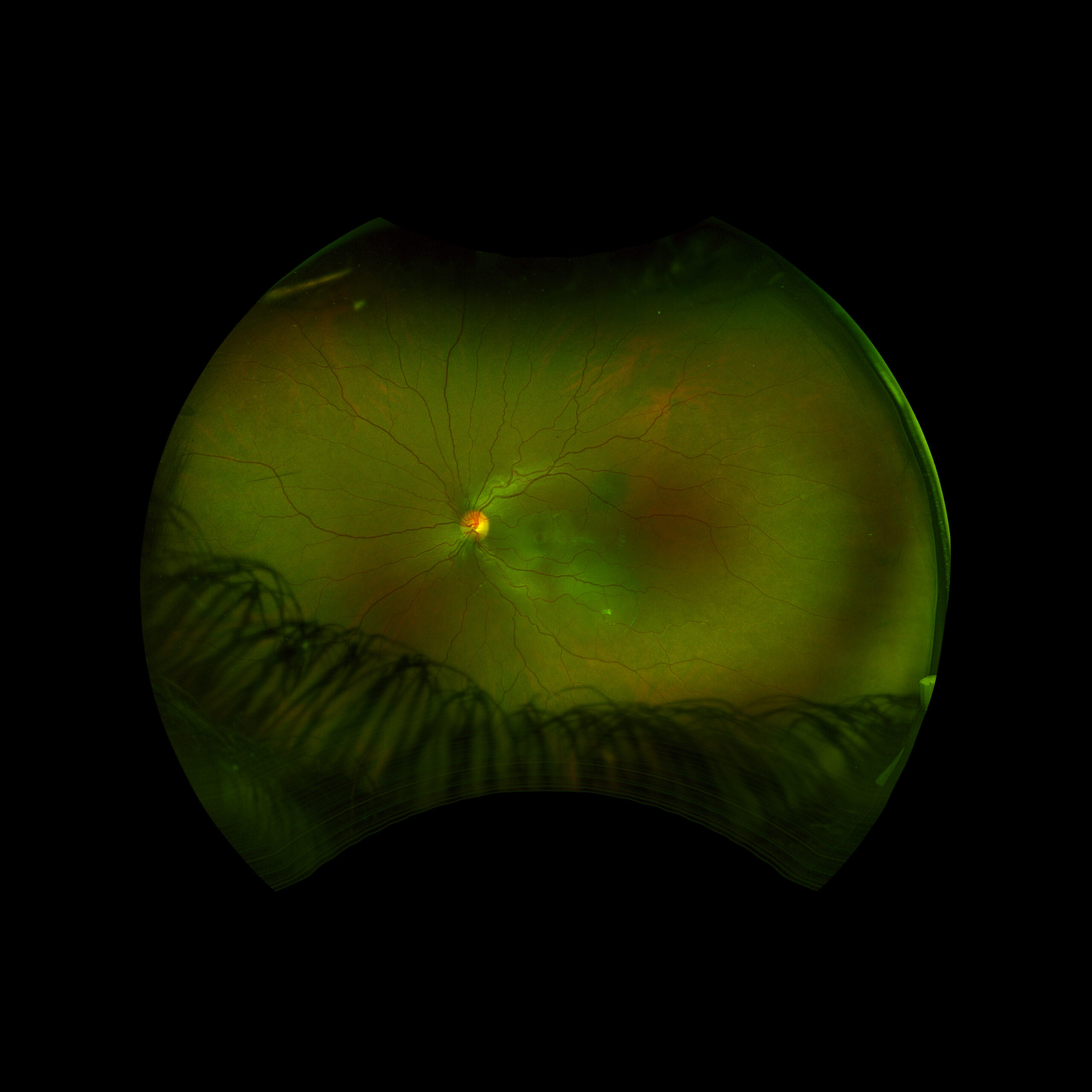
Figure 1: Wide field fundus photo of the left eye showing subretinal fluid and a hypo-pigmented choroidal elevation centrally and inferotemporally. Note an unrelated choroidal nevus along the superotemporal arcade.
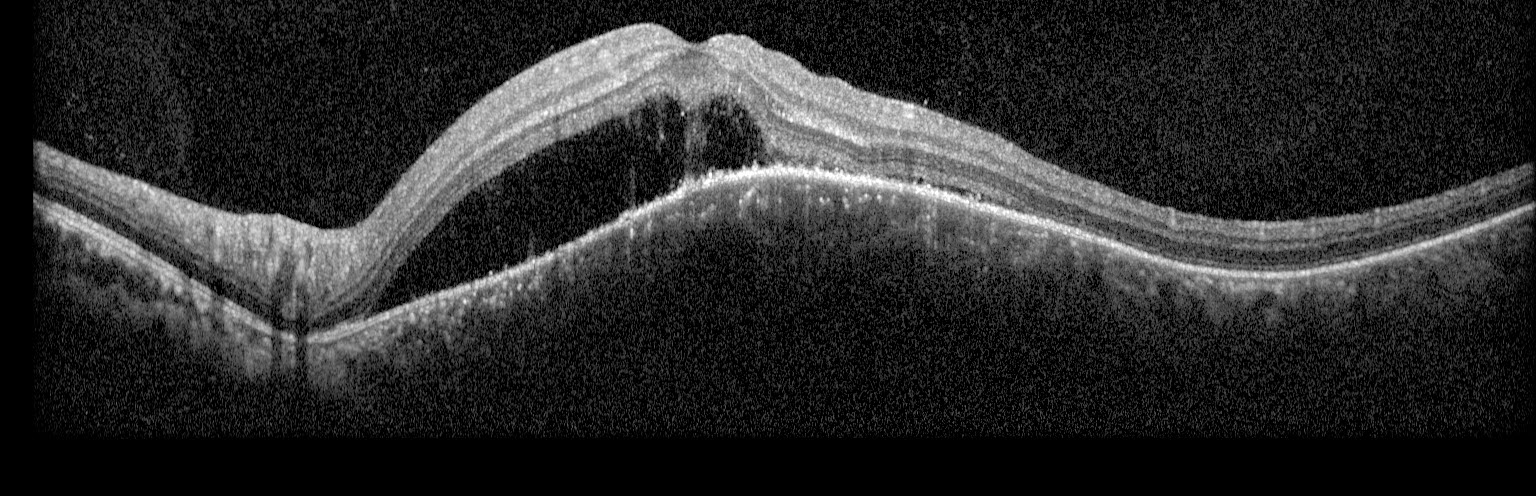
Figure 2: Optical coherence tomography (OCT) of the left eye with subretinal fluid and choroidal elevation.
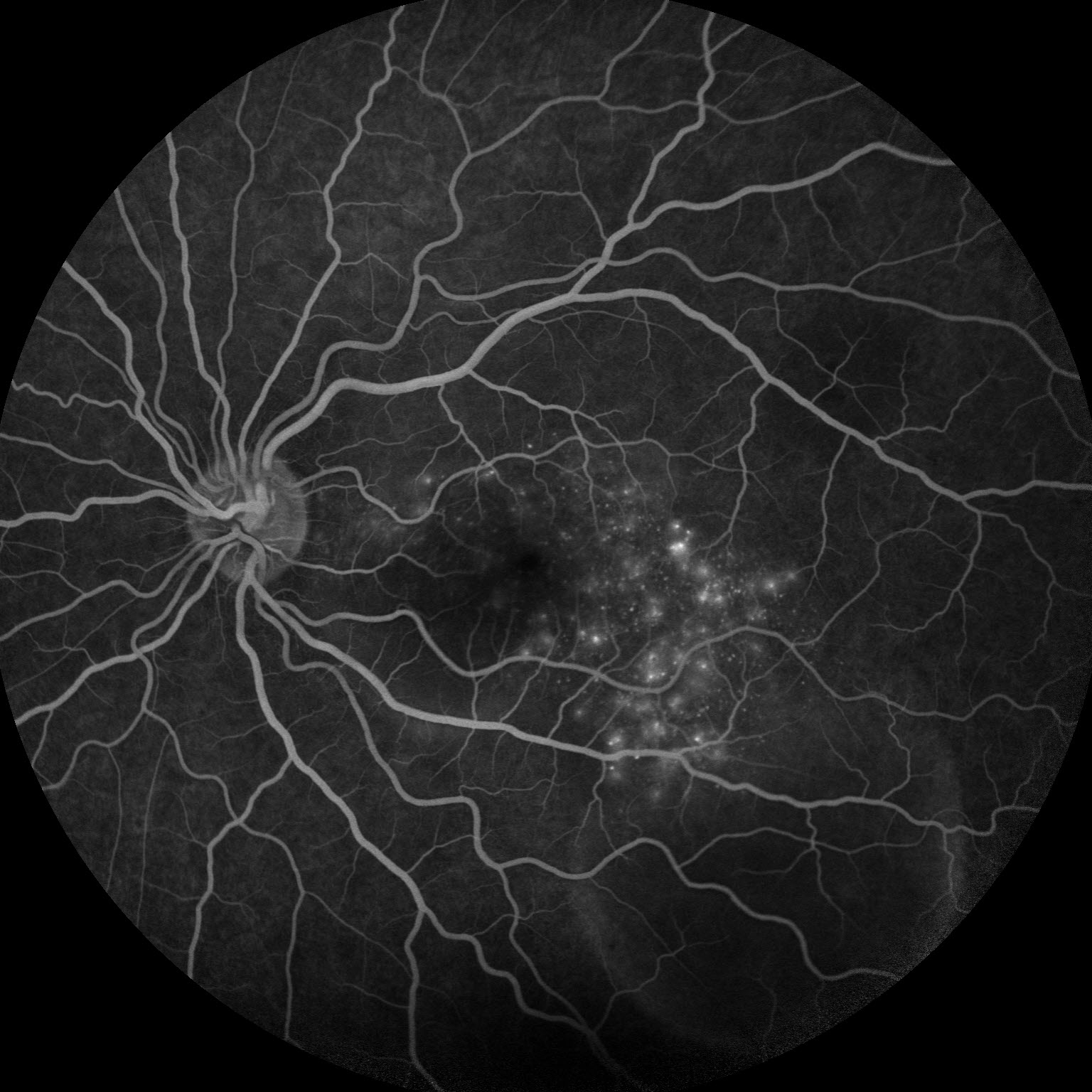
Figure 3: Fluorescein angiography (FA) of the left eye at 07:20 demonstrating hyperfluorescent punctate spots overlying the inferotemperal lesion.
Description of Case:
An 18 year old female of Mexican descent presented after waking up with a headache in the frontal region and behind the left eye. Subsequently, she noted an enlarging dark spot in her left eye’s vision. She denied any previous ocular history. Medical history was notable for class IV obesity and asthma.
Right eye exam was unremarkable. Left eye exam was notable for rare pigmented vitreous cell, subretinal fluid, as well as a hypo-pigmented choroidal elevation centrally and inferotemporally, confirmed on optical coherence tomography (OCT) (Fig 1 &2). Fluorescein angiography (FA) demonstrated late hyperfluorescent punctate spots overlying the inferotemperal lesion (Fig 3). B scan confirmed choroidal thickening without a discrete mass.
Early Vogt-Koyanagi-Harada was suspected, with the differential also including non-pigmented choroidal tumor infectious etiology. Labs to rule out infectious etiology were negative. Treatment was initiated with a prednisone taper, and the patient’s visual acuity, subretinal fluid, and choroidal elevation were greatly improved the following week, supporting the diagnosis. The patient went on to develop recurrences in subsequent years, requiring further prednisone tapers and the addition of immunomodulatory therapy (methotrexate).
Overview:
Vogt-Koyanagi-Harada (VHK) disease is characterized by bilateral granulomatous panuveitis and systemic manifestations related to T-cell mediated autoimmune attacks of melanocytes in the eyes, ears, meninges, skin, and hair.1 It is most commonly seen in darkly pigmented populations (especially individuals of Asian, Hispanic, and Middle Eastern descent), in women, and in younger adults.2,3 In the United States, it is a rare cause of uveitis, making up only 3-4% of referrals to tertiary care centers.4 Clinically, VKH disease presents in four different phases: prodromal, acute uveitic, convalescent, and chronic recurrent.
The prodromal phase includes mostly extraocular manifestations, such as headache, flu-like symptoms, meningismus, tinnitus, and hearing loss.5
Days to weeks later, features of acute uveitis manifest suddenly and bilaterally, beginning with the posterior segment and potentially spreading to the vitreous and anterior segment if untreated.1 Posterior pole signs include areas of subretinal fluid and choroidal thickening, leading to breakdown of the retinal pigment epithelium and the disease’s characteristic serous retinal detachments.1
In the convalescent stage, inflammation subsides, leaving behind a depigmented fundus often described as a “sunset glow.”3
Some patients develop chronic recurrences of granulomatous anterior uveitis that are resistant to steroids and can involve a myriad of complications, including cataract, subretinal fibrosis, chorioretinal atrophy, choroidal neovascular membrane, and glaucoma.6
Systemic manifestations:
Besides the ophthalmic symptoms and complications, patients with VKH disease can often develop systemic findings related to autoimmune targeting of melanocyte antigens. Signs and symptoms include depigmentation of the eyebrows, eyelashes, and scalp, alopecia, poliosis, CSF pleocytosis, headache, and hearing manifestations such as sensorineural hearing loss, tinnitus, dysacusis, and a sensation of fullness in the ear.1,7
Workup and Diagnosis:
The diagnosis of VKH is based on clinical findings as above in combination with the absence of penetrating trauma or surgery to the eye or a more likely diagnosis based on clinical and laboratory findings, with ancillary investigations commonly including fluorescein angiography (FA), indocyanine green angiography (ICGA), optical coherence tomography (OCT), ultrasonography, electroretinography (ERG), and cerebrospinal fluid analysis.3
Treatment:
Early treatment with high-dose systemic corticosteroids is crucial to manage the acute stage of VKH, with slow tapering to prevent recurrence.5 More recent data has shown better outcomes with the addition of local steroid injections and/or implants to treat refractory ocular symptoms, and immunomodulatory therapy (IMT) to treat systemic manifestations while sparing patients from complications of systemic steroids in the long-term.1,7
Prognosis:
The visual prognosis of VKH depends heavily on the stage in which it presents. Early treatment in the acute stage is associated with a good visual prognosis and even cure, while chronic recurrent disease is often refractory to treatment and prone to complications such as choroidal neovascular membrane, glaucoma, cataract, and chorioretinal atrophy.6
Summary of the Case:
Vogt-Koyanagi-Harada (VHK) disease is a form of granulomatous panuveitis that targets melanocytes. In the acute stage, posterior uveitis with subretinal fluid and choroidal thickening predominates along with hearing, neurologic, and integumentary changes. The chronic stage is characterized by anterior uveitic recurrences and ocular complications. Early control with high-dose steroids and possibly IMT is critical for an optimal prognosis.
References:
- O’Keefe GAD, Rao NA. Vogt-Koyanagi-Harada disease. Surv Ophthalmol. 2017;62(1):1-25. doi:10.1016/j.survophthal.2016.05.002
- Read RW, Holland GN, Rao NA, et al. Revised Diagnostic Criteria for Vogt-Koyanagi-Harada Disease: Report of an International Committee on Nomenclature. Am J Ophthalmol. 2001;131(5).
- Tayal A, Daigavane S, Gupta N. Vogt-Koyanagi-Harada Disease: A Narrative Review. Cureus. Published online April 23, 2024. doi:10.7759/cureus.58867
- Ohno S, Char DH, Kimura SJ, Richard O’Connor G. Vogt-Koyanagi-Harada Syndrome. Am J Ophthalmol. 1977;83(5):735-740. doi:10.1016/0002-9394(77)90142-8
- Lodhi S, Reddy L, Perum V. Clinical spectrum and management options in Vogt–Koyanagi–Harada disease. Clin Ophthalmol. 2017;Volume 11:1399-1406. doi:10.2147/OPTH.S134977
- Urzua CA, Herbort C, Valenzuela RA, et al. Initial-onset acute and chronic recurrent stages are two distinctive courses of Vogt-Koyanagi-Harada disease. J Ophthalmic Inflamm Infect. 2020;10(1):23. doi:10.1186/s12348-020-00214-2
- Stern EM, Nataneli N. Vogt-Koyanagi-Harada Syndrome. In: StatPearls. StatPearls Publishing; 2024. Accessed August 6, 2024. http://www.ncbi.nlm.nih.gov/books/NBK574571/
Faculty Approval by: Akbar Shakoor, MD
Copyright statement: Copyright Taylor Johnson, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Uveitic Cataract in Biopsy-proven Sarcoid-associated Uveitis
Home / Intraocular Inflammation and Uveitis / Complications of Uveitis
Title: Uveitic Cataract in Biopsy-proven Sarcoid-associated Uveitis
Authors: Amanda Wade, BA; Marissa Larochelle, MD
Date: 07/18/2024
Keywords/Main Subjects: uveitis, cataracts, uveitic cataract, uveitis complications, ocular sarcoidosis
Diagnosis: Cataract secondary to uveitis
Description of Case: A 46-year-old woman with biopsy-proven sarcoid-associated uveitis complicated by bilateral peripapillary and macular CNVM (choroidal neovascular membrane) with submacular fibrosis presented with gradually decreasing vision over the previous year in the left eye.
Initial ocular presentation occurred approximately 20 years prior with bilateral eye pain and redness (anterior sarcoid uveitis). She was followed for “macular degeneration” at the time due to posterior involvement appreciated on exam and received laser (PDT) over the course of multiple visits. She then transitioned care to another physician who proceeded with Avastin injections for about 1 year. She eventually began methotrexate for systemic control of her sarcoidosis.
Figure 1. Fundus photos:
Peripapillary and macular CNVM pictured in both right and left eyes; notably clear view of the fundus bilaterally.
Left eye
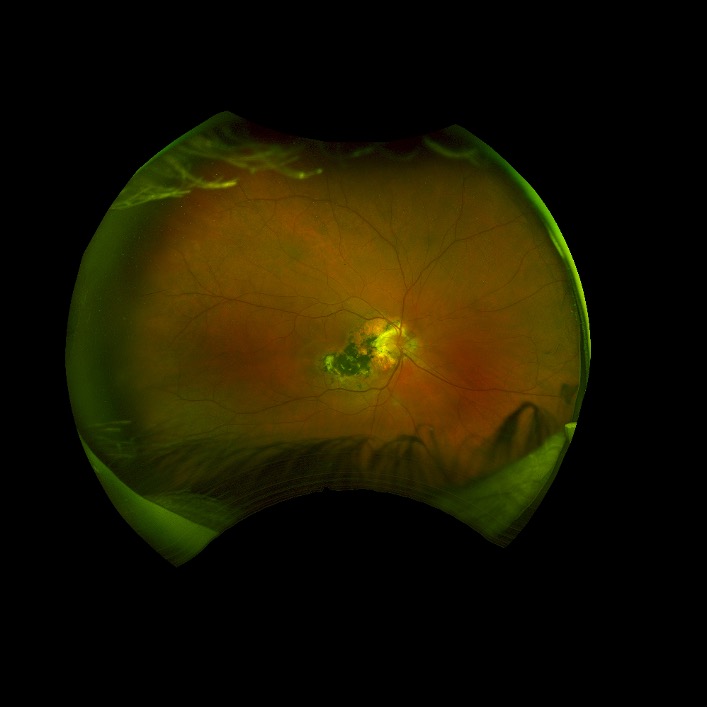
Right eye
Figure 2. Macula OCT – left eye:
Sub retinal hyperreflectivity, intraretinal hyperreflectivity, multiple intraretinal cysts over chorioretinal scarring
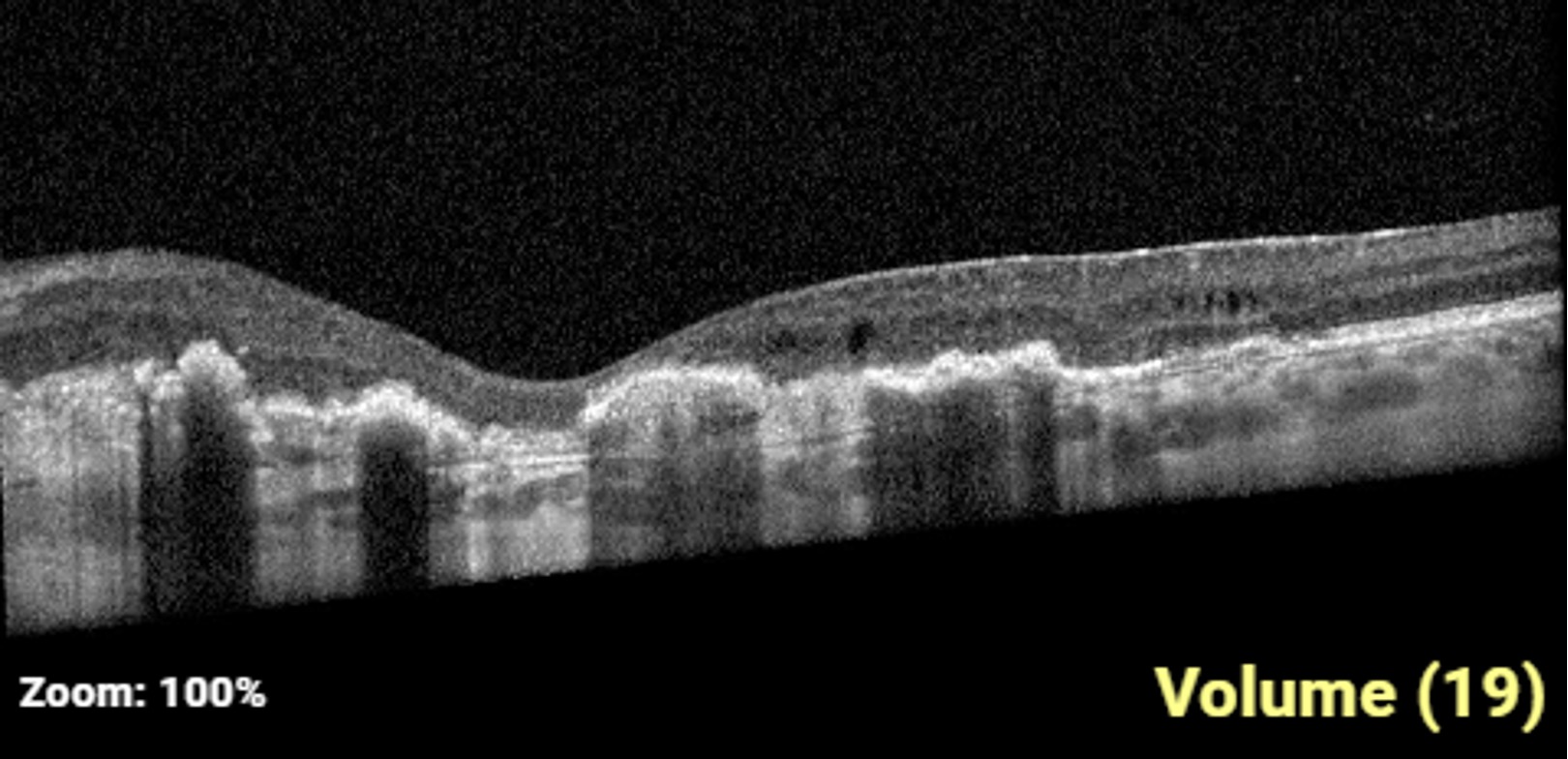
She had a recurrence of anterior uveitis in left eye which was treated with topical steroids. Due to ongoing uveitis and recurrent bilateral CME (cystoid macular edema), she warranted advancement of her systemic immunomodulatory therapy and Humira was initiated. The patient also received an OZURDEX® (dexamethasone intravitreal implant) in the left eye which led to almost complete resolution of IRF (intraretinal fluid) in the left eye.
It was hypothesized that some of the IRF might be non-modifiable due to degenerative cysts over areas of atrophy with ERM (epiretinal membrane).
Over subsequent visits, her visual acuity in the left eye worsened from her baseline of 20/80 OS to 20/150 OS (cc). Prior to OZURDEX® injection, her exam was notable for a 1+ noncentral posterior subcapsular cataract (PSC) in the left eye. Over the next year, this progressed to a 2+ central PSC with anterior cortical changes.
Figure 3. Left eye fundus photo 10 months after dexamethasone intravitreal implant.
While previous retinal findings are still visible, note the central PSC obstructing the visual axis and additional anterior cortical spoking.
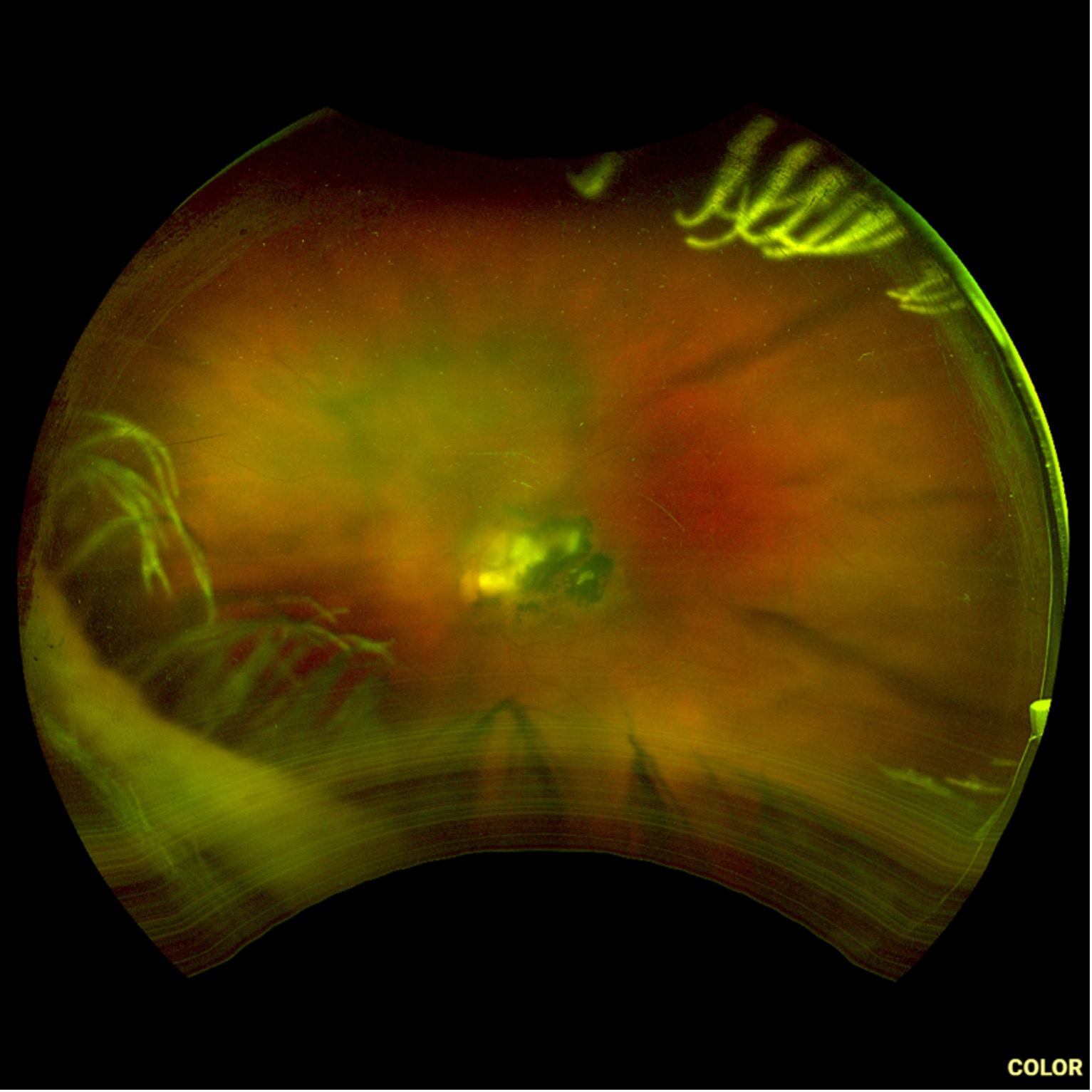
After ruling out macular etiologies of her visual decline (macula OCT stable from previous), she underwent cataract surgery with intraocular lens placement in the left eye, which resulted in an improved VA from 20/150 (mr) to 20/80 OS sc on post-op day 1.
Discussion:
Cataract formation can be accelerated due to myriad causes, including inflammatory processes like uveitis (specifically ocular sarcoidosis in the above patient’s case). Both the disease (uveitis) as well as a mainstay of its treatment (steroids) lead to cataract formation in patients with uveitis. Because the lens material is a crystalline structure that is avascular, nutrition and maintenance of the lens are derived from intraocular fluid contents. It follows that intraocular inflammation, and inflammatory cytokines, disrupt the metabolism of the lens through diffusion, particularly posteriorly. This is largely due to the lack of endothelial support at the back of the lens. Cataracts in eyes with uveitis require a different set of considerations when approaching a surgical decision.
Diagnosis:
The most common symptoms of age-related cataracts are decreasing visual acuity/clarity both at distance and near, and glare and halos, particularly at night; however, diagnosis of a uveitic cataract must be secondary to a confirmed case of uveitis and is more likely to present with a PSC that creates visually significant changes. Uveitic cataracts may be surgically addressed if they obscure posterior segment pathology and preclude appropriate monitoring of disease activity or less urgently when interfering with the patient’s visual function.
Management/Considerations:
When approaching the uveitic cataract, considerations must be made to account for infectious versus inflammatory uveitis. In cases of uveitis due to viral etiologies, such as HSV or VZV anterior uveitis or acute retinal necrosis, it is prudent to increase systemic antivirals to therapeutic levels in the peri-operative period. In the case of sarcoid uveitis and other chronic ocular inflammatory diseases, having an eye without active inflammation for 3 months is recommended prior to cataract surgery. This may be achieved with an appropriate immunomodulatory regimen.
Inflammation control:
To mitigate the risk for inflammatory flares and complications both pre- and postoperatively, steroids are considered standard of care. Topical eye drops (prednisolone acetate 1%), oral prednisone and periocular or intraocular steroids are often used individually and in combination to maximally decrease the risk of a uveitis flare in the perioperative period.
Surgical control/challenges:
Both the size of the surgical incision and the capsulorhexis should be of sufficient length/diameter to prevent iris prolapse and decrease the risk of capsular phimosis and posterior synechiae respectively. An in-the-bag lens placement has been highly preferred as it reduces the chances of subsequent inflammation, although ongoing research suggests that sulcus placement might have advantages. Small pupils, anterior or posterior synechiae, and zonular dehiscence, which can all be a result of uveitis, increase the risk of zonular dehiscence, anterior chamber hemorrhage and pigment dispersion and must be approached with care. Additionally, the anterior capsule can often be fibrotic and can require additional manipulation during capsulorhexis and cause tension on the zonules.
Postoperative considerations:
In addition to inflammation control, due to the use of steroids coupled with an intraocular surgery, intraocular pressure should be monitored closely for both pressure spikes and low pressures that may be concerning for ciliary body failure, ciliary effusion, cyclitic membranes or early phthisis. Other complications in the post-operative period can include the formation of a posterior capsular opacity (PCO), cystoid macular edema (CME), and epiretinal membrane formation (ERM), among others.
Format: Case Report
References:
- Shaw E, Patel BC. Complicated Cataract. [Updated 2023 Jul 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK572139/
- Liu YC, Wilkins M, Kim T, Malyugin B, Mehta JS. Cataracts. Lancet. 2017 Aug 5;390(10094):600-612. doi: 10.1016/S0140-6736(17)30544-5. Epub 2017 Feb 25. PMID: 28242111.
- Alekseev BN, Sibai SA. Opredelenie termina “oslozhnennye katarakty” [Definition of the term “complicated cataracts”]. Vestn Oftalmol. 1996 Sep-Oct;112(4):26-7. Russian. PMID: 9019908.
- Avetisov SE, Razumova IY, Avetisov KS. Rezul’taty khirurgicheskogo lecheniya oslozhnennoi uveal’noi katarakty [Results of surgical treatment of complicated uveal cataract]. Vestn Oftalmol. 2020;136(5. Vyp. 2):209-213. Russian. doi: 10.17116/oftalma2020136052209. PMID: 33063966.
- Avetisov SE, Razumova IY, Avetisov KS. Rezul’taty khirurgicheskogo lecheniya oslozhnennoi uveal’noi katarakty [Results of surgical treatment of complicated uveal cataract]. Vestn Oftalmol. 2020;136(5. Vyp. 2):209-213. Russian. doi: 10.17116/oftalma2020136052209. PMID: 33063966.
Faculty Approval by: Marissa Larochelle, MD
Copyright statement: Copyright Amanda Wade, Marissa Larochelle, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/