


Terson Syndrome
Home / Retina and Vitreous / Other Retinal Vascular Diseases
Title: Terson Syndrome
Author (s): Dan Nguyen, MS4
Photographer: Unknown
Date: 8/16/17
Keywords/Main Subjects: Terson Syndrome, boat hemorrhage
Diagnosis: Terson Syndrome
Description of Case: Terson Syndrome is intraocular hemorrhage (IOH) in the presence of intracranial hemorrhage or elevated intracranial pressure (ICP). IOH can include vitreous, sub-hyaloid, pre-retinal, intraretinal, and subretinal bleeding. Vitreous hemorrhage is the most frequent subtype of ocular hemorrhage in Terson syndrome. It may present with visual loss and can cause permanent blindness if severe. It is most commonly associated with subarachnoid hemorrhage (SAH) secondary to an anterior circulation aneurysmal rupture. It can be seen in up to 46% of SAH patients, and its presence is associated with a higher mortality in SAH. The sudden spike in ICP with aneurysmal rupture is thought to underlie the cause of IOH. This increased pressure is transmitted along the optic nerve sheath and causes sudden intraocular venous HTN and rupture of thin capillary walls. The diagnosis is made with a fundoscopic exam, and ophthalmological exams should be routinely performed in patients diagnosed with SAH. Related complications of Terson Syndrome include macular holes, epiretinal membrane formation, proliferative vitreoretinopathy, retinal folds, retinal detachments, and optic neuropathy.
Some recommendations in literature suggest 3-6 months of observation after the acute event followed by vitrectomy if there is no improvement in visual acuity. But other studies suggest that earlier intervention before 3 months is optimal, especially in younger patients.
The fundus photos are from patient TS, a 35 year old female who was seen at Moran emergently for papilledema. Previously, she was found to have a Chiari malformation and had a suboccipital craniectomy at the end of May. On 7/29 she woke up on the floor, unable to move for 25 minutes, and had bilateral vision loss and was only able to see colors and shapes. At Moran she was noted to have stage III papilledema and peripapillary hemorrhages and was sent to the University ED. She had an LP with an opening pressure of 35 cm and was admitted to Neurosurgery with a shunt placed on 7/31. She was seen in the Neuro-Ophthalmology clinic on 8/16 with continued vision impairment, particularly in her right eye. On exam, her VA was 20/200 OD and 20/25 OS with correction. She had a tilt afferent pupillary defect OD. Slit lamp showed vitreous RBCs bilaterally, OD > OS. Fundoscopic exam (see pictures below) showed large pre-retinal/subhyaloid hemorrhages OU, more central OD vs OS. Visual fields showed mild central scotoma OD. The etiology of her ICP elevation is uncertain. She was treated with a pars plana vitrectomy (PPV) with membrane peeling (MP) in her right eye. Following surgery her VA improved to 20/30 post-op day 1.
Images or video:

Fundoscopic photograph demonstrating boat shaped retinal heme in the right eye of patient TS due to Terson Syndrome. In this specific case, the heme is a result of pre-retinal hemorrhage, posterior to the internal limiting membrane and anterior to the nerve fiber layer. Sub-hyaloid hemorrhage, a result of heme between the posterior vitreous base and the internal limiting membrane, presents similarly as a boat shaped retinal heme. The only way to reliably differentiate between sub-hyaloid and pre-retinal heme is during surgery.

Fundoscopic photograph demonstrating boat shaped heme in OS of patient TS from either sub-hyaloid or pre-retinal hemorrhage. Here, the macula is less affected vs OD, and this is seen clinically with her VA, 20/200 OD and 20/25 OS.
Summary of the Case: TS is a 35 YO F who presents with decreased vision secondary to pre-retinal hemorrhage due to transient elevated ICP resulting in intra-ocular hemorrhage. Intra-ocular hemorrhage subsequent to intracranial hemorrhages or elevated IOP is known as Terson Syndrome. The patient was treated with PPV with MP and her vision OD improved from 20/200 pre-surgery to 20/30 post-surgery.
Format: image
References:
- Hassan, A. Giuseppe, L. Eelco, FM et al. Terson’s Syndrome. Neurocrit Care. 2011 May;15:554-558.
- Fang, K. Knox, DL. The Ocular Pathology of Terson’s Syndrome. American Academy of Ophthalmology. 2010;117:1423-1429.
- Garweg, JG. Koerner, F. Outcome Indicators for Vitrectomy in Terson Syndrome. Acta Ophthalmologica. 2009;87:222-226.
Faculty Approval by: Paul Berstein, MD
Copyright statement: Copyright Dan Nguyen, ©2015. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Identifier: Moran_CORE_24577
Fundus Photography and Fluorescein Angiography of Branch Retinal Artery Occlusion
Home / Retina and Vitreous / Other Retinal Vascular Disease
Title: Fundus Photography and Fluorescein Angiography of Branch Retinal Artery Occlusion
Author: Johnny Lippincott
Photographer: Unknown
Date: 8/28/2017
Images:


Keywords/Main Subjects: Branch Retinal Artery Occlusion, BRAO, Retinal Vascular Disease
CORE Category:
Disorders of the Retina and Vitreous: Other Retinal Vascular Diseases, 8: Arterial Occlusive Disease
Diagnosis: Branch Retinal Artery Occlusion
Description of Image: Branch Retinal Artery Occlusion (BRAO) is the obstruction of adequate bloodflow to a retinal artery distal to the central retinal artery. Hypoperfusion of downstream tissue leads to retinal ischemia, cell damage, and vision loss. More than two-thirds of BRAOs are caused by emboli composed of either cholesterol, platelet-fibrin, or calcific plaques.1 Cholesterol emboli are known as Hollenhurst plaques. Emboli tend to occlude vessels at points of bifurcation. BRAOs are almost always found in the temporal, not nasal, arteries.2
Patients typically present with monocular sudden, painless vision loss in a sectoral or altitudinal distribution. Severity of vision loss (in field and acuity) varies significantly and depends on the area affected and degree of ischemia. 80% of affected eyes recover visual acuity of 20/40 or better.3 Given this rate of recovery, invasive therapy is usually avoided in cases without significant foveal involvement. Ocular massage or paracentesis can dislodge an embolus (albeit rarely), and employing laser therapy to disrupt an embolus may improve visual outcomes but is associated with vitreous hemorrhages.2, 4
This fundus photograph demonstrates characteristic findings in BRAO, including a sectoral area of opaque (“cloudy”), whitened, edematous retina. Retinal veins form the boundaries of this ischemic area, which here includes part of the superior fovea. A cotton-wool spot has developed (arrow). Though not seen in this photograph, an embolus is visible on fundus photography in a majority of cases. Fluoroscein angiography displays a hyperfluorescent spot (arrow) that may represent the culprit embolus that has migrated beyond the point of occlusion. The area of ischemia is hypofluorescent.
References:
- Hayreh SS, Zimmerman MB. Fundus changes in branch retinal arteriolar occlusion. Retina. 2015 Oct 1;35(10):2060-6.
- Yanoff M, Duker JS. Retinal arterial obstruction. Ophthalmology 4th edition. Elsevier Saunders. 2014;129.
- Brown GC, Magargal LE, Shields JA, Goldberg RE, Walsh PN. Retinal arterial obstruction in children and young adults. Ophthalmology. 1981 Jan 1;88(1):18-25.
- Man V, Hecht I, Talitman M, Hilely A, Midlij M, Burgansky-Eliash Z, Achiron A. Treatment of retinal artery occlusion using transluminal Nd: YAG laser: a systematic review and meta-analysis. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2017:1-9.
Faculty Approval by: Griffin Jardine, MD
Copyright statement: Copyright Johnny Lippincott, ©2016. For further information regarding the rights to this collection, please visit: https://morancore.utah.edu/terms-of-use/
Disclosure (Financial or other): The author has no financial or business interests that may be affected by this site or its contents.
Identifier: Moran_CORE_24567
Fundus Photography, Fluorescein Angiography, and Indocyanine Green Angiography of Tuberculous Choroiditis
Home / Intraocular Inflammation and Uveitis / Infectious Uveitis
Author (s): Kristen Russell
Photographer: N/A
Date: 11/1/2016
Image or video:

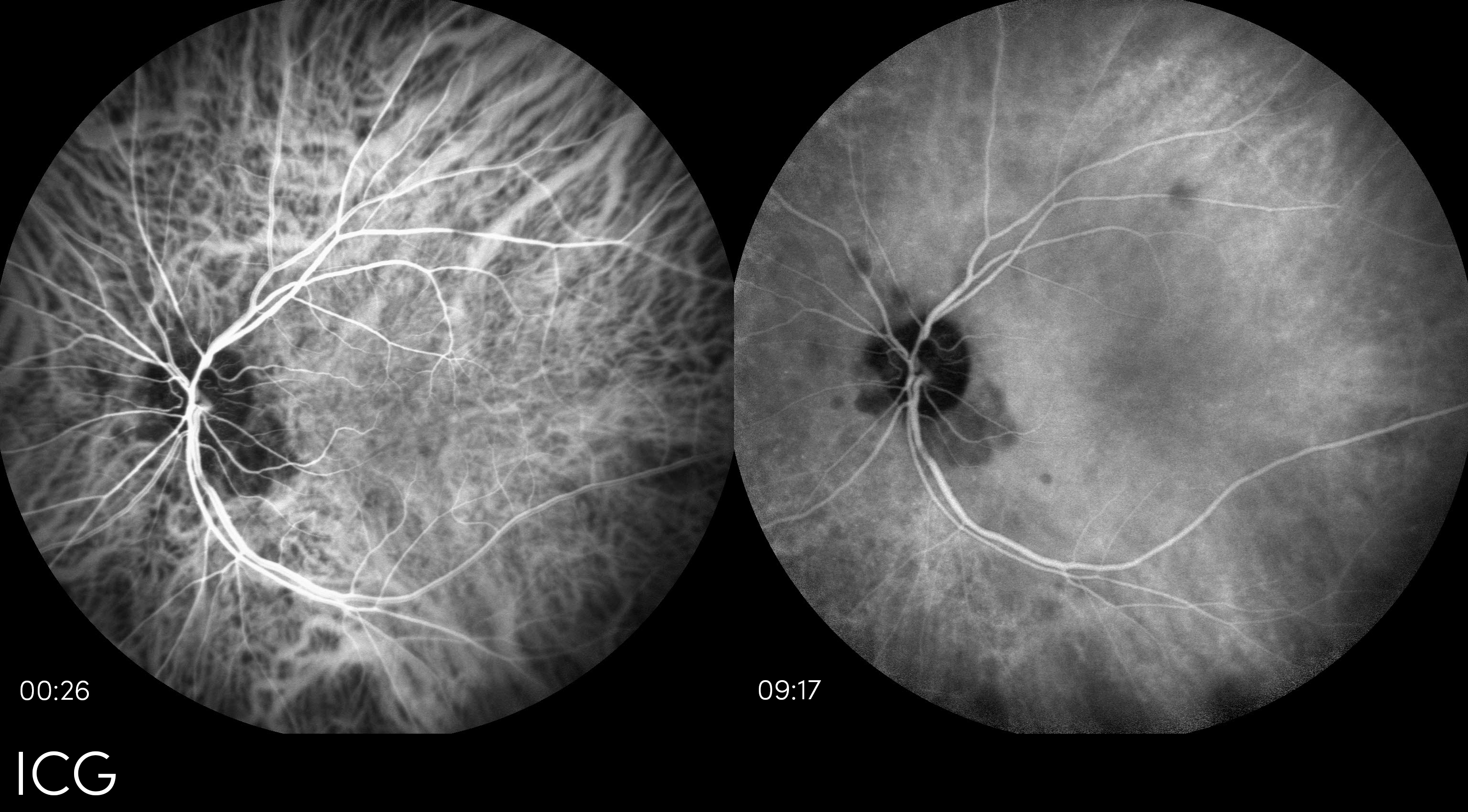

Keywords/Main Subjects: Tuberculous Choroiditis
Diagnosis: Tuberculous Choroiditis
Description of Image:
Here we are showing the fundus photo, FA, and ICG from a 51 year old female. She was referred to uveitis clinic because her BCVA was 20/60 OS and she had a history of recurrent vitreous cell, floaters, and photophobia. Previously, she had several PPD’s (purified protein derivative) that were positive, however, her chest x-ray had remained negative. On exam, her right eye was normal, but her left eye had mutton fat keratic precipitates, posterior synechiae, 1-2+ cell in the anterior chamber, vitreous haze, loss of the foveal reflex and inferior punched out chorioretinal lesions. Her left fundus photo showed mild haze and intraretinal heme. On FA, there were numerous nasal lesions that demonstrated early hypofluorescence and late hyperfluorescence. The ICG had multiple hypo-fluorescent lesions which matched the FA. Her laboratory work-up revealed a positive QuantiFERON-TB Gold. She was started on ethambutol, isoniazid, pyrazinamide, and rifampin and within one month, BCVA returned to 20/30 OS.
Mycobacterium Tuberculosis can cause a variety of ocular manifestations ranging from retinitis to scleritis. However, the most common is focal or multifocal choroiditis. Patients often present with no history of active TB and only half have a positive chest x-ray (1). Instead, patients may report photopsias, floaters, or scotomas. On slit lamp exam, there will be signs of uveitis, such as mutton fat keratic precipitates, cell and flare, posterior synechiae, and vitreous cells. Fundus exam may reveal areas of atrophy or active choroidal lesions. On fluorescein angiography (FA), the active site will typically show early hypo-fluorescence but late hyperfluorescence—versus indocyanine green angiography (ICG), where the lesions remain hypo-fluorescent throughout the course of the study. The differential diagnosis for these symptoms and imaging studies includes syphilis, sarcoidosis, birdshot retinochoroidopathy, and Vogt-Koyanagi-Harada syndrome. A common work-up, therefore, might include a QuantiFERON-TB Gold, RPR, FTA-ABS, chest x-ray, ACE, HLA-A29, hearing screening, and lumbar puncture. If a patient is diagnosed with ocular tuberculosis, the treatment is anti-TB medication, sometimes in conjunction with systemic corticosteroids.
References:
- Bennett, J. E., Dolin, R., Blaser, M. J., Mandell, G. L., & Douglas, R. G. (2015). Infectious Causes of Uveitis. In Mandell, Douglas, and Bennetts Principles and practice of infectious diseases. (pp 1423-1431). Elsevier Inc.
- American Academy of Ophthalmology. (n.d.). White Dot Syndromes: Multifocal Choroiditis and Panuveitis. Retrieved from https://www.aao.org/focalpointssnippetdetail.aspx
- Gupta, A., Bansal, R., & Gupta, V. (2014). Tuberculosis, Leprosy, and Brucellosis. In Ophthalmology (pp. 716-719). Elsevier Inc.
Faculty Approval by: Griffin Jardine, MD
Copyright statement: Copyright Author Name, ©2017. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
Identifier: Moran_CORE_
Disclosure (Financial or other): None
Anterior Basement Membrane Dystrophy (ABMD)
Home / External Disease and Cornea / Corneal Dystrophies and Ectasias
Title: Anterior Basement Membrane Dystrophy (ABMD)
Authors: Paul D Chamberlain, BS, Brian Zaugg, MD
Photographer: Becky Weeks, CRA
Date: 8/14/2017
Image or video:

Slit lamp photograph of the characteristic map lines seen in ABMD.

Topography of an eye with ABMD showing irregular astigmatism with superior flattening of the central cornea.

Topography of the same eye 6 weeks following superficial keratectomy with diamond burr polishing.
Keywords/Main Subjects: Anterior basement membrane dystrophy; Corneal dystrophy; recurrent erosions;
Diagnosis: Anterior Basement Membrane Dystrophy (ABMD) (also: Epithelial basement membrane dystrophy(EBMD), map-dot-fingerprint dystrophy, Cogan’s microcystic epithelial dystrophy).
Description of Image:
The first corneal topography shown here (image 2a) is from a patient who initially presented for evaluation of worsening vision in the right eye (20/60 with correction) and was found to have visually significant cataracts. As part of his pre-operative evaluation this corneal topography was taken, showing a very irregular cornea. During his follow-up visit he reported that since the initial evaluation he had experienced right eye pain upon wakening in the morning, consistent with a recurrent erosion. The scheduled cataract surgery was changed to superficial keratectomy with diamond burr polishing following which his vision corrected to 20/20 in the right eye, with a subsequent correction of the abnormal topography (image 2b) . Thus, in patients presenting with visual loss who are found to have both ABMD and cataracts, the ABMD should be treated first.
Anterior basement membrane dystrophy (ABMD) is a disease affecting the basement membrane of the corneal epithelial cells and is the most common corneal dystrophy. It is commonly bilateral though can be asymmetric. Patients may present with significant visual impairment due to ABMD, or it may be an incidental finding upon corneal examination. ABMD may also present as recurrent epithelial erosions. In this case the patient may complain of intermittent, severe eye pain, usually upon awakening in the morning and opening their eyes.
Diagnosis of ABMD is made from physical examination of the cornea (see image 1). The characteristic findings include either thick, irregular lines that may resemble a coastline (maps); small, punctate gray-white opacities with distinct edges (dots or microcysts); or thin hair-like lines arranged in parallel (fingerprints). These represent changes in the corneal epithelial basement membrane. Corneal topography may demonstrate irregularities in the cornea (see image 2a).
Mild cases without recurrent erosions may be treated with lubricating ointment and hypertonic saline drops/ointment. Recurrent erosions may require bandage contact lenses and antibiotic ointment. If these fail to provide benefit for the patient, or if the ABMD is causing significant visual impairment either superficial keratectomy with diamond burr polishing or photorefractive keratectomy might be required to relieve symptoms. Both treatments have been found to be highly successful, especially in the treatment of recurrent erosions.
References:
- Laibson PR. Recurrent corneal erosions and epithelial basement membrane dystrophy. Eye and Contact Lens: Science and Clinical Practice. 2010; 36(5):315-7.
- Wong VW, Chi SC, Lam DS. Diamond burr polishing for recurrent corneal erosions: results from a prospective randomized controlled trial. Cornea. 2009; 28(2):152-156.
- Sridhar MS, Rapuano CJ, Cosar CB, Cohen EJ, Laibson PR. Phototherapeutic keratectomy versus diamond burr polishing of Bowman’s membrane in the treatment of recurrent corneal erosions associated with anterior basement membrane dystrophy. Ophthalmology. 2002; 109(4):674-679.
Faculty Approval by: Brian Zaugg, MD; Griffin Jardine, MD
Copyright statement: Moran Eye Center, ©2017. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE.
Identifier: Moran_CORE_24507
Disclosure (Financial or other): None
Fundus Photography of Traumatic Choroid Rupture with Angioid Streaks
Home / Retina and Vitreous / Retinal Degenerations Associated with Systemic Disease
Title: Fundus Photography of Traumatic Choroid Rupture with Angioid Streaks
Author: Alaina Hamilton, B.S.
Photographer: James Gilman, CRA, FOPS
Date: June 2004 (image 1) and October 2013 (image 2)
Image or video:
June 2004

October 2013
Keywords/Main Subjects: Angioid Streaks; Traumatic Choroid Rupture;
Secondary CORE Category:
Retina and Vitreous / Other Retinal Vascular Diseases
Retina and Vitreous / Posterior Segment Manifestations of Trauma
Diagnosis: Traumatic Choroid Rupture with Angioid Streaks
Description of Image:
The fundus photos shown are from a middle-age man with pseudoxanthoma elasticum and angioid streaks that presented to the clinic a month after he was struck as a pedestrian by a motor vehicle. The first image shows his right eye before the accident and the second image was taken a month after the accident. The image taken before the accident demonstrates peau d’orange (arrow), which is the pebbly orange appearance to the retina and a classic finding seen with angioid streaks. On exam after the accident, he was found to have traumatic choroid rupture and extensive subretinal hemorrhage in both eyes, the left worse than the right. His vision after the accident was found to be 20/70 in the right eye and count fingers in the left (prior to the accident he had 20/20 vision in both eyes). A couple of months after this visit, he returned to clinic where his vision had improved to 20/25 in the right eye and 20/40. However, he was found to have developed a choroidal neovascular membrane in the left eye so he was treated with intravitreal anti-VEGF at that time. His vision in his left eye improved to 20/25 with the treatment and he is still currently stable with the monthly anti-VEGF injections.
Angioid streaks are bilateral, irregular tapering lines that lie deep to the retina and radiate from the peripapillary region within the posterior pole. Later ingrowth of fibrovascular tissue from the choroid into the sub-retinal pigment epithelial space can partially or totally obscure the streaks margins. The streaks represent breaks in a weakened Bruch’s membrane. These breaks can occur spontaneously or may be the result of even mild trauma. Other complications that can arise include choroidal neovascularization and subretinal hemorrhage. Approximately 50% of patients with angioid streaks have an associated systemic disease, pseudoxanthoma elasticum being the most common. Other commonly associated diseases can be remembered with the mnemonic PEPSI: Pseudoxanthoma elasticum, Ehler-Danlos syndrome, Paget’s disease of bone, Sickle cell disease, and Idiopathic, although there are many other less common associations. Diagnosis is usually made by clinical examination alone but can be confirmed with fluorescein angiography. Fluorescein angiography is particularly useful in evaluating choroidal neovascularization, as this would warrant treatment. Intravitreal anti-VEGF therapy can be used to stabilize vision if CNV has occurred, but often requires repeated injections due to recurrence.
References:
Mathew R, Sivaprasad S, Augsburger JJ, Corrêa ZM. Retina. Vaughan & Asbury’s General Ophthalmology, 19e New York, NY: McGraw-Hill
Georgalas I, Papaconstantinou D, Koutsandrea C, et al. Angioid streaks, clinical course, complications, and current therapeutic management. Therapeutics and Clinical Risk Management. 2009;5:81-89.
Faculty Approval by: P.S. Bernstein, MD, PhD
Copyright statement: Copyright Author Name, ©2016. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
Identifier: Moran_CORE_24493
Disclosure (Financial or other): None
Central Optic Opacification of a Polymethyl Methacrylate Intraocular Lens Consistent with Snowflake Degeneration
Home / Lens and Cataract / Complications of Cataract Surgery
Title: Central Optic Opacification of a Polymethyl Methacrylate Intraocular Lens Consistent with Snowflake Degeneration
Author: Bryan Masino, BS
Photographer: David Chang, MD, Liliana Werner, MD, PhD
Date: 09/07/2016
Keywords/Main Subjects: Intraocular Lens (IOL), Snowflake Degeneration, Polymethyl Methacrylate (PMMA)
Diagnosis: Three-piece PMMA IOL with clinically significant optic opacity due to snowflake degeneration.
Description of Case: The Polymethyl Methacrylate (PMMA) intraocular lens (IOL) was implanted in the left eye of a 53-year-old female patient in 1985. The lens exhibited slow, yet progressive optic opacification that eventually led to decreased visual acuity. It was therefore explanted on October 13, 2009 (Figure 1), nearly 25 years after the implantation date, by Dr. David Chang from Los Altos, CA. The explanted lens was sent to the Intermountain Ocular Research Center (Mamalis/Werner Laboratory) for further analysis.

Figure 1: Surgical view of eye with snowflake degeneration of a PMMA IOL.
Gross examination in the dry state revealed that the optic displayed a prominent white discoloration located within the central 3 mm (Figure 2). Microscopic examination of the dry lens showed multiple lesions with an aspect characteristic of snowflake degeneration (Figure 3).1-3 These lesions were found where the central optic opacity was seen during clinical and gross examinations, and were distributed within the optic substance at different depths. The lesions had the appearance of small spherical “crystals” that were bounded by an outer pseudocapsule suggestively composed of compressed and degenerated PMMA.
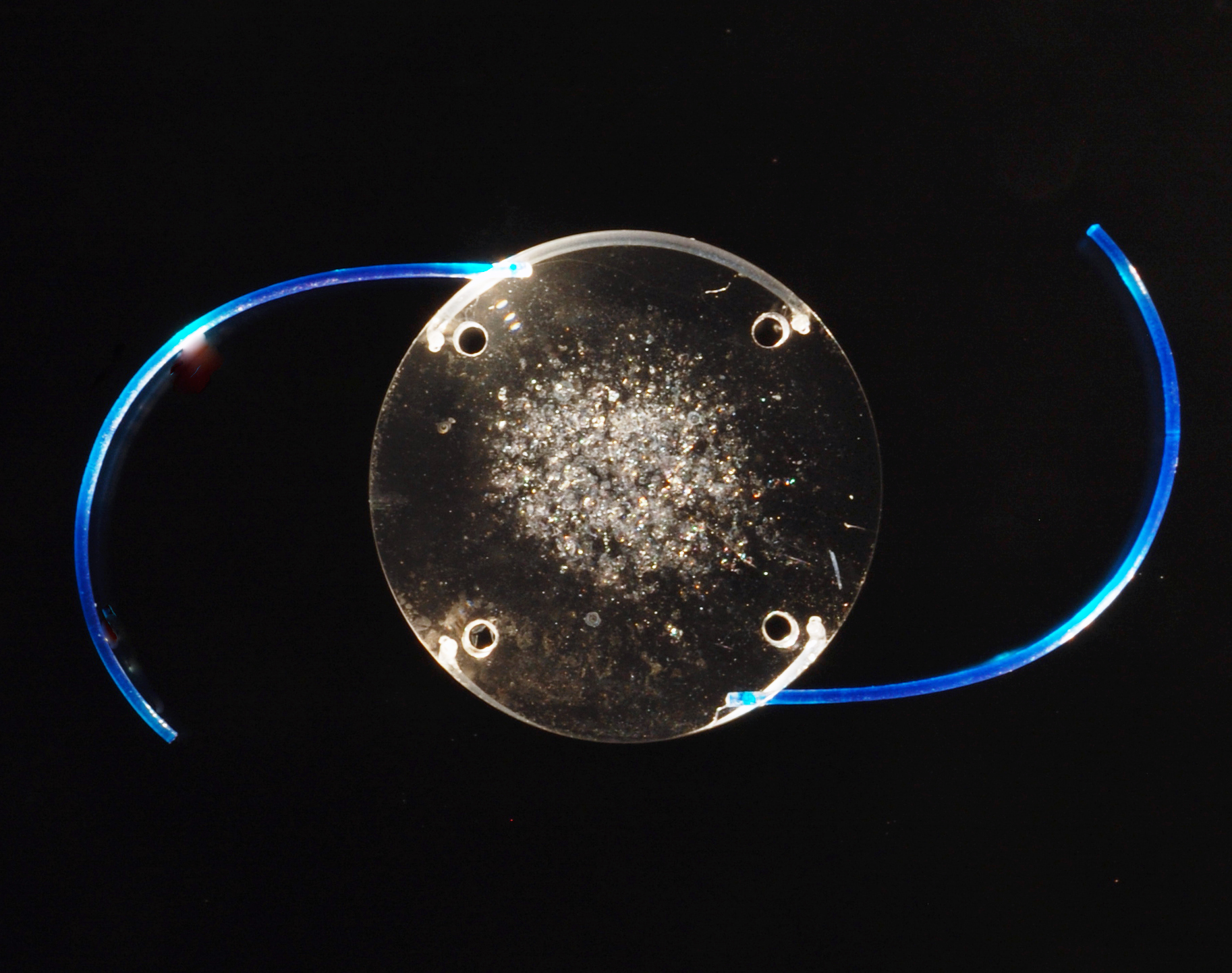
Figure 2: Gross photograph of the explanted lens in the dry state.

Figure 3: Light photomicrograph of the dry lens (X400)
The explanted lens was evaluated under anterior segment optical coherent tomography (AS-OCT),4 which showed the abnormal lesions within the optic substance (Figure 4).
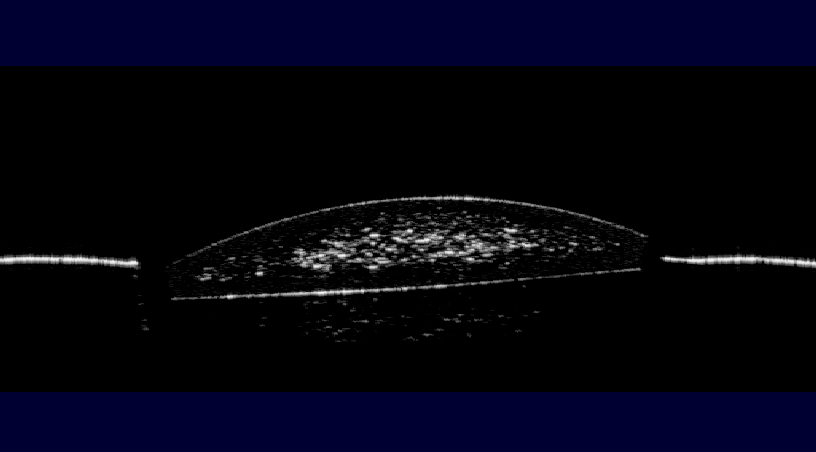
Figure 4: AS-OCT photograph of the dry lens.
As to mimic the environment in which the lens resided while inside of the patients eye, the explanted PMMA IOL was hydrated in balanced salt solution (BSS) and once again analyzed under gross and light microscopy, as well as under the AS-OCT. Upon gross examination the severity of the lens opacification was markedly increased and the diameter thereof nearly doubled to that of approximately 5mm (Figure 5). AS-OCT revealed a much denser and widespread area of snowflake lesions.

Figure 5: Gross photograph of the explanted lens in the hydrated state.
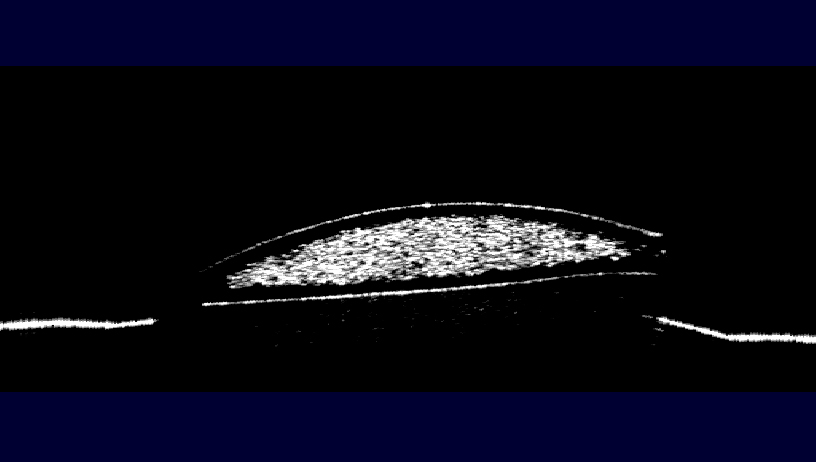
Figure 6: AS-OCT photograph of the lens after hydration.
The hydrated explant was also evaluated under Scheimpflug photography with densitometry analysis to assess back light scattering.5 This revealed a very high light scattering with a density of 225 CCT at the level of the intraoptical lesions (Figure 7).

Figure 7: Light scattering analysis of the lens under Scheimpflug photography.
Light transmittance was also assessed using a spectrophotometer,5 showing decreased light transmittance through the explanted lens when compared to a control (Figure 8).
Figure 8: Light transmittance analysis by spectrophotometer of the explanted lens and a control PMMA IOL.
The microscopic appearance of the intraoptical lesions is consistent with snowflake degeneration of PMMA material. This is a slowly progressive opacification of PMMA lenses, occurring sometimes 10 years or more after implantation. Snowflake degeneration has been observed in three-piece PMMA lenses implanted between the early 1980’s and the mid 1990’s, which were generally manufactured by injection molding. It has been hypothesized that this degeneration is a result of long-term ultraviolet (UV) exposure. The explanted lenses analyzed in our laboratory had spherical lesions, which were interpreted as foci of degenerated PMMA material clustered in the central zone and midperipheral portion of the optic. This led to the hypothesis that the central optic was exposed to UV light over an extended period, whereas the peripheral optic may be protected by the iris. Therefore, snowflake lesions are generally not observed in the optic periphery. Although the snowflake lesions are dry, it has been observed that an unusual amount of water is collected within the affected optic area upon hydration of explanted PMMA lenses with this condition, leading to more significant optic opacification. Consequently, the clinical significance of snowflake degeneration may depend on the amount of water collected within the IOL optic.3 In vitro studies on three-piece PMMA lenses explanted because of this condition showed that they are associated with a considerable increase in forward light scatter, as well as with a decline of optical quality/performance indicators.6 This can give rise to a variety of subjective complaints, including glare in various illuminating conditions, halos around bright lights, color and contrast loss, as well as hazy vision. These symptoms may prompt IOL explantation even in the absence of significant decrease in visual acuity. It is important to point out that today industry has far greater control over the production of the PMMA material/lenses. Modern PMMA lenses are mostly produced by lathing; therefore, we do not expect to see development of snowflake degeneration in association with them.
Summary of the Case: The patient experienced a decrease in visual acuity due to slow-onset progressive opacification of a PMMA IOL that was implanted over 20 years ago. After the necessary explantation surgery, the lens was examined and exhibited marked snowflake degeneration, which has been observed with three-piece PMMA IOLs that were manufactured during the early 1980’s and the mid 1990’s.
References:
- Werner L. Causes of intraocular lens opacification or discoloration. J Cataract Refract Surg 2007; 33:713-26.
- Apple DJ, Peng Q, Arthur SN, et al. Snowflake degeneration of polymethyl methacrylate posterior chamber intraocular lens optic material: a newly described clinical condition caused by unexpected late opacification of polymethyl methacrylate. Ophthalmology 2002; 109:1666-75.
- Dahle N, Werner L, Fry L, Mamalis N. Localized, central optic snowflake degeneration of a PMMA intraocular lens: Clinical report with pathological correlation. Arch Ophthalmol 2006; 124:1350-3.
- Werner L, Michelson J, Ollerton A, Leishman L, Bodnar Z. Anterior segment optical coherence tomography in the assessment of postoperative intraocular lens optic changes. J Cataract Refract Surg 2012; 38:1077-85.
- Michelson J, Werner L, Ollerton A, Leishman L, Bodnar Z. Light scattering and light transmittance in intraocular lenses explanted because of optic opacification. J Cataract Refract Surg 2012; 38:1476-85.
- Werner L, Stover JC, Schwiegerling J, Das KK. Effects of intraocular lens opacification on light scatter, stray light, and overall optical quality/performance. Invest Ophthalmol Vis Sci 2016; 57(7):3239-47.
Faculty Approval by: Liliana Werner, MD, PhD, and Nick Mamalis, MD.
Identifier: Moran_CORE_24223
Copyright statement: Copyright 2017. Please see terms of use page for more information.
Disclosure (Financial or other): The author does not have any financial interest in the material and methods mentioned.
Photographic Essay of Idiopathic Intracranial Hypertension-Associated Choroidal Neovascular Membranes
Home / Retina and Vitreous / Choroidal Disease
Title: Photographic Essay of Idiopathic Intracranial Hypertension-Associated Choroidal Neovascular Membranes
Authors: Christopher D. Conrady, MD, PhD, Anastasia Neufeld, MD, M.E. Hartnett, MD, Kathleen Digre, MD
Date: 07/25/2017
Keywords/Main Subjects: Idiopathic intracranial hypertension; Choroidal neovascular membrane; Optic nerve swelling
Secondary CORE Category: Home / Neuro-Ophthalmology / Causes of Decreased Vision
Diagnosis: Choroidal neovascular membrane secondary to chronic optic disc swelling
Description of Case: BG is a 13-year-old, right-handed girl with past medical history of Hashimoto’s thyroiditis who developed acute blurring of the vision in the right eye while reading a book at school. She was seen by outside providers who noted bilateral disc edema and referred her to the emergency department for further evaluation. At time of presentation, she denied pulsatile tinnitus, transient visual obscurations, headache, and diplopia. She denied any new medications but did endorse a 50 lb. weight gain.
On examination, visual acuity was count fingers at 4’ in the right eye and 20/20 in the left. Intraocular pressure was 22 mmHg and 20 mmHg in the right and left eye respectively. There was a small (0.3-0.6 log units), right-sided afferent pupillary defect .Hardy-Rand-Ritter color plates were significantly depressed in the right but not left eye. Flicker fusion was severely depressed at 9 Hz in the right and
normal in the left at 31 Hz. Anterior segment was unremarkable. Dilated eye examination noted significant peripapillary hemorrhages and 360 degrees of edema of the optic nerve in the right eye (Figure 1). The left eye was notable for stage II disc edema.
A Humphrey visual field of the left eye noted an enlarged blind spot, while a Goldman visual field noted a large central scotoma of the right eye (Figure 1c-d). Fluorescein angiography noted a leaking lesion consistent with a peripapillary choroidal neovascular membrane (Figure 2a-c) with fluid extending into the fovea (Figure 2e). B-scan of the globes did not identify optic nerve head drusen (Figure 2d).
The patient was referred to the retina service that performed an intravitreal avastin injection in the right eye. Within a few months, vision returned in the right eye to 20/40 with significant improvement of cecocentral scotoma and resolution of the subretinal fluid (Figure 3a-e).
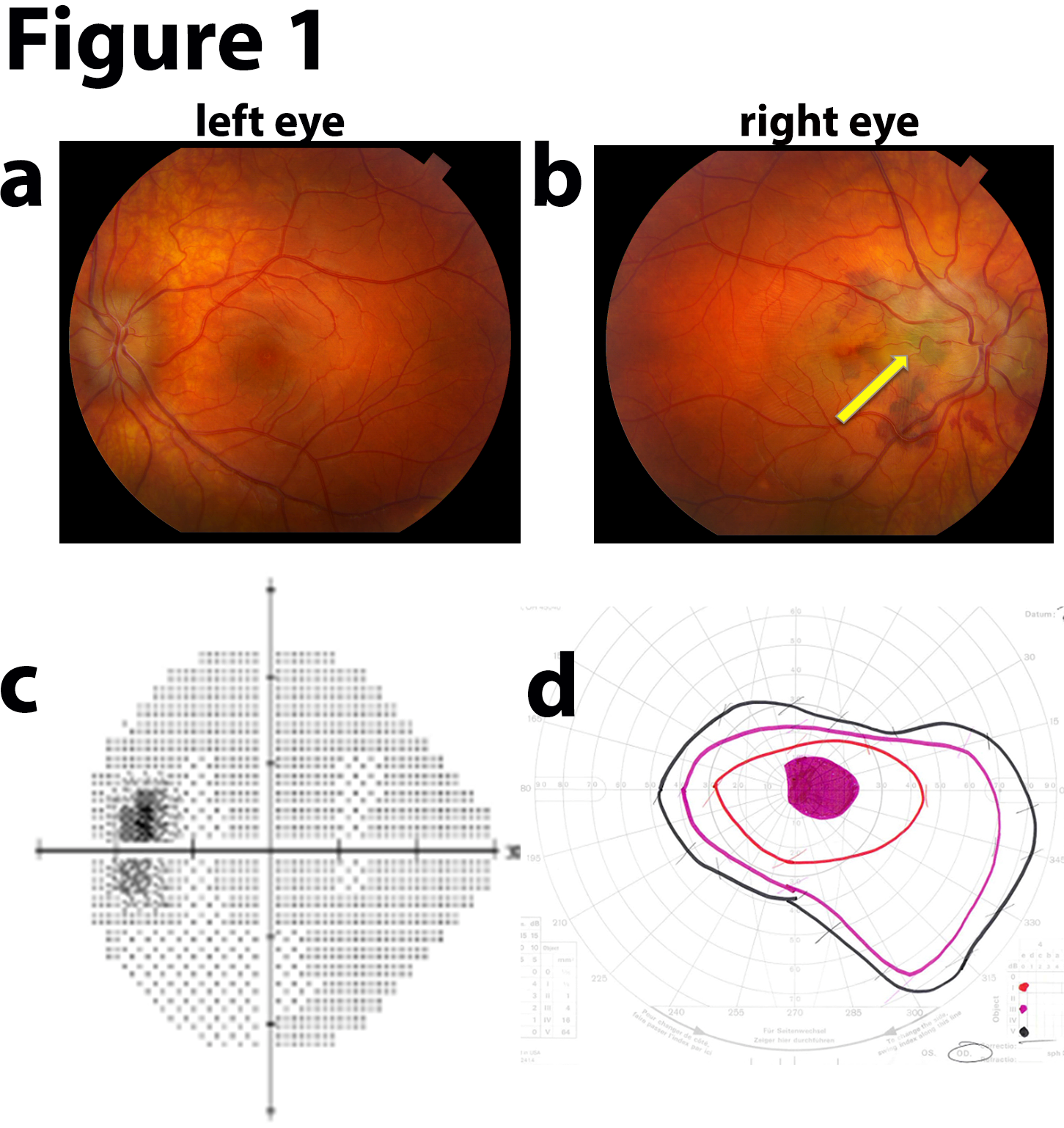
Figure 1: Acute vision loss in the right eye with bilateral disc edema.
(a-b) Fundus photographs of the left and right eye showed bilateral disc edema. In the right eye (b), there is peripapillary subretinal blood and a choroidal neovascular membrane (yellow arrow). (c) Humphrey visual field noted an enlarged blind spot in the left eye. (d) Goldman visual field noted a central scotoma of the right eye. Left column, left eye; right column, right eye.

Figure 2: Choroidal neovascular membrane with surrounding subretinal fluid and no signs of optic nerve head drusen of the right eye.
(a-c) Fluorescein angiogram of the right eye noted a peripapillary lesion that leaked late consistent with a choroidal neovascular membrane (red arrows). (d) There were no signs of optic nerve head drusen on ultrasound. (e) Macular OCT identified subretinal fluid extending from the optic nerve into the macula (yellow arrow).

Figure 3: Resolution of subretinal fluid with improvement in vision and visual field in the right eye.
(a) Follow up macular OCT of the right eye noted resolution of subretinal fluid in the right eye. Optic disc edema (b-c) and visual fields were also improving in both eyes (d-e). Left column, left eye; right column, right eye.
Summary of the Case:
- Choroidal neovascular membranes can complicate longstanding optic nerve edema in children.
- These membranes respond very well to anti-VEGF therapy.
Faculty Approval by: Dr. Kathleen Digre
Identifier: Moran_CORE_24138
Copyright statement: Copyright Conrady, ©2017. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Birdshot Chorioretinopathy
Home / Ophthalmic Pathology / Retina and Retinal Pigment Epithelium
Title: Birdshot Chorioretinopathy, Case Report
Author: Taylor Fields, Fourth Year Medical Student, Medical College of Georgia
Photographers: Cyrie Fry, CRA, and Paula Morris, CRA.
Date: 08/23/15
Secondary CORE Category: Retina and Vitreous / Focal and Diffuse Choroidal and Retinal Inflammation
Keywords/Main Subject: Posterior Uveitis; Chorioretinopathy; Birdshot; BSCR; BCR Diagnosis- Birdshot Chorioretinopathy
Brief Description of Case:
This case is a 52-year-old white female who presented complaining of “hundreds of floaters” that had been progressing for several months in both eyes. She noted a crescent shape to the floaters and a one month of a small blurry spot in the inferonasal visual field in her right eye. She also described nyctalopia, a less vivid quality to her vision and morning photopsias and black spots upon awakening that lasted about 30 minutes.
Visual acuity was 20/20 in each eye and intraocular pressure by tonopen was 21 and 19 in the right and left eyes respectively. Her pupillary exam, visual fields, color vision and motility were all normal. The Fundus exam revealed 1+ disc edema, bilateral epiretinal membranes, narrowing of her retinal vessels and numerous mid-peripheral white/yellow choroidal spots bilaterally (Pictures 1-4). ICG photos revealed areas of decreased dye perfusion within the choroid (Picture 3).
The differential diagnosis included inflammatory uveitis (birdshot chorioretinopathy, sarcoidosis), infectious causes (TB, syphilis, Lyme disease) and masquerade syndromes such as choroidal lymphoma. She was found to be HLA-A29 positive. An extensive laboratory work-up was otherwise negative. Full-field electroretinograms (ffERGs) showed attenuated scotopic dim blue flash ERGs to about 59% with slowed photopic b-wave time of 35 milliseconds.
Diagnosis
The patient was diagnosed with birdshot chorioretinopathy (BSCR). Her fundus exam and ffERG results are typical of early stages of BSCR. This disease is an idiopathic posterior uveitis that commonly presents in Caucasians in their sixth decade of life. It has a strong association with the human leukocyte antigen HLA-A29 (96% of patients are carriers).1,2 BSCR typically presents with a gradual centripetal loss of vision and presence of floaters.3
Treatment
The patient was started on a 60 mg prednisone taper as well as mycophenolate and cyclosporine. Systemic immunomodulatory therapy is the mainstay of treatment for BSCR. Concomitant therapy with an antimetabolite and a T-cell inhibitor can be beneficial for preserving vision, and decreasing the cumulative glucocorticoid dose.4,5
Prognosis
BSCR is considered a chronic and progressive isolated ocular disorder. A majority of patients develop retinal dysfunction; however, central vision is typically spared until late in the disease. Visual acuity at initial presentation is a prognostic factor for long-term visual outcomes with appropriate treatment.6
Images

Picture 1. Full color montage photos of the right (A) and left (B) eyes. These show characteristic pisiform lesions of BSCR within the periphery of the retina (black arrows).
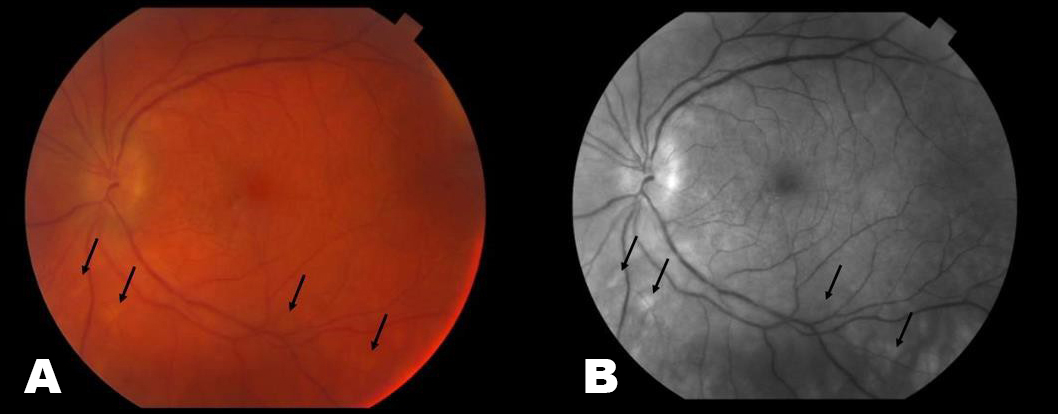
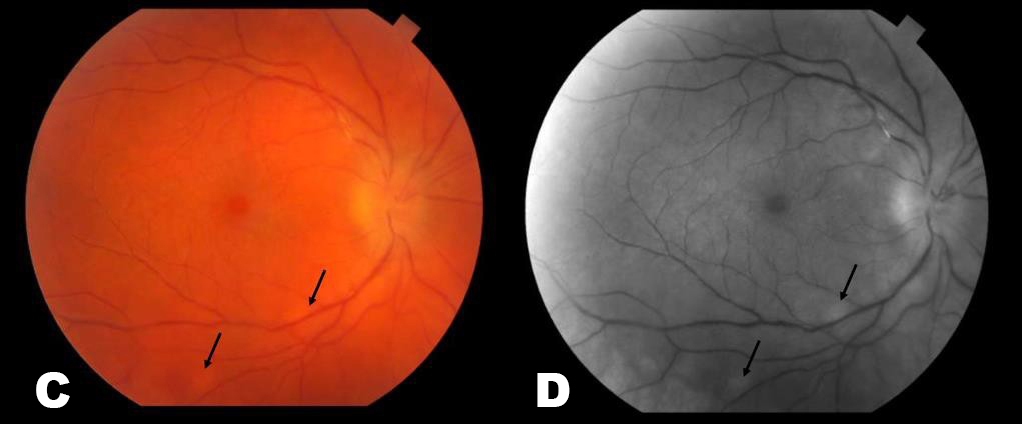
Picture 2. These are color (A & C) and red free (B & D) photos of the left (A & B) and right (C & D) fundi. The periphery has areas of white/yellow choroidal spots (black arrows). There is also narrowing of vessels and disc edema present.
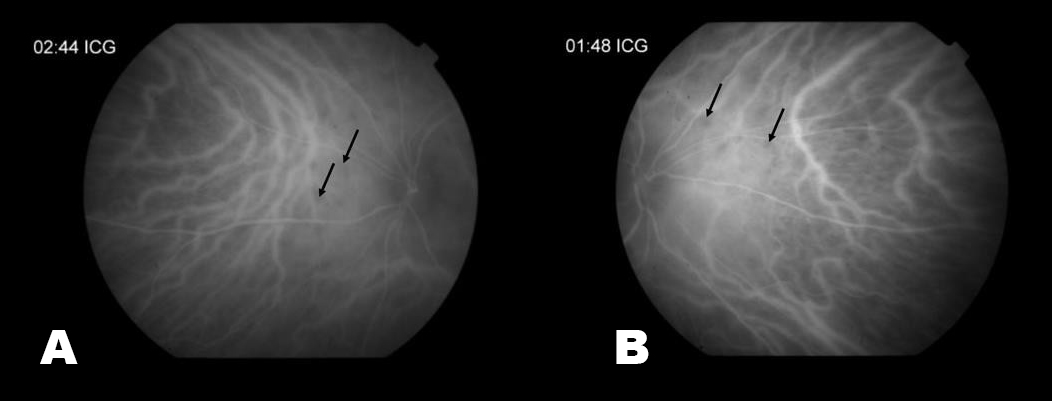
Picture 3. These are late ICG photos of the nasal portions of the Left (A) and Right (B) retinas. There is decreased choroidal perfusion, and thus dark spots, in the areas of inflammation (black arrows).

Picture 4. These are wide field fluorescein angiograms (FA) of the right (A) and left (B) fundi. This shows vasculature leakage, periphlebitis, and disc edema.
Summary of Case
This is a 52 year-old white female diagnosed with birdshot chorioretinopathy who presented with several months of worsening floaters and was found to have the characteristic midperipheral white/yellow choroidal lesions bilaterally. She was found to be HLA-A29 positive, confirmatory of the diagnosis.
References
Levinson RD, Brezin A, Rothova A, et al. Research criteria for the diagnosis of birdshot chorioretinopathy: results of an international consensus Am J Ophthalmol 2006; 141:185 – 187.
- Brézin AP, Monnet D, Cohen JH, Levinson HLA-A29 and birdshot chorioretinopathy.Ocul Immunol Inflamm. 2011;19(6):397-400.
- Shah KH, Levinson RD, Yu F, et al. Birdshot chorioretinopathy. Surv Ophthalmol 2005; 50:519 –
- Becker MD, Wertheim MS, Smith JR, Rosenbaum Long-term follow-up of patients with birdshot retinochoroidopathy treated with systemic immunosuppression. Ocul Immunol Inflamm 2005; 13:289–293.st
- Kiss, Szilard, Ahmed, Muna, Letko, Erik, & Foster, Stephen. (2005). Long-term Follow- up of Patients with Birdshot Retinochoroidopathy Treated with Corticosteroid-Sparing Systemic Immunomodulatory Therapy. Ophthalmology, 112(6), 1066-1071.e2.
- Tomkins-Netzer O, Taylor SR, Lightman Long-term clinical and anatomic outcome of birdshot chorioretinopathy. JAMA Ophthalmol 2014; 132:57 – 62.
Faculty Approval by: Griffin Jardine, MD
Identifier: Moran_CORE_23873
Opthalmoplegic Migraine/Recurrent Painful Ophthalmoplegic Neuropathy
Home / Neuro-Ophthalmology / Evaluation of Diplopia
Title: Opthalmoplegic Migraine/Recurrent Painful Ophthalmoplegic Neuropathy
Author Information: Ryan O’Meilia MS4, University of Oklahoma; Judith Warner MD, John A. Moran Eye Center
Financial Disclosure: None
Date: September 2015
Keywords: Opthalmoplegic Migraine; Recurrent Painful Ophthalmoplegic neuropathy
Description:
Chief Complaint: Lid drooping and drifting eye
History of Present Illness:
This case discusses a 3 year old girl with a symptomatic, recurring oculomotor palsy. Her first episode was at age one but completely resolved. Four days prior to her most recent presentation she had a gastroenteritis with vomiting and decreased energy. Her parents did not note a fever, upper respiratory infection, cough, eye pain, rash or HA in the past few weeks. The gastroenteritis resolved two days later but around that same time she developed right ptosis. Over the next two days the eyelid drooping progressed and the patient developed deviated eye and asymmetric pupils.
Her previous episode of oculomotor nerve palsy developed much like the present one. The parents report a sub-acute onset over a few days that progressed from ptosis, to anisocoria and strabismus. The episode resolved without treatment over about one week. An MRI was performed at an outside hospital and the read was as follows: “Enhancement of the right third cranial nerve (CN) which is most prominent immediately adjacent to the origin of the nerve at the brain stem. The enhancement extends anteriorly along the nerve to the cavernous sinus.” Her clinical presentation and imaging were felt to be consistent with inflammatory neuritis, most likely post viral.
Past Medical History: otherwise non-contributory
Past Surgical History: None
Medications: None
Allergies: No known drug allergies
Immunizations: Up to date
Development: Age appropriate
Family History: No family history of eye disease, migraine, MS, neurologic disease, childhood cancers. Maternal cousins with celiac disease and DM1
Social History: Lives with parents; 2 cats and a dog. Attends daycare
Review of Symptoms: All negative except as above
Ocular Physical Exam:
- General: Alert and oriented, NAD
- Visual Acuity: Fix and Follow OU
- Pupils
- OD: 5mm > 4mm
- OS: 4mm > 2mm
- EOM
- Primary gaze: right eye deviated inferiorly and temporally
- OD:
- -3 adduction
- -2 elevation
- -1 depression
- -0 abduction
- OS:
- Penlight
- Full EOM
- Orbits: No deformities, ecchymosis or edema
- Lids/Lashes: Right ptosis, MRD1 5mm, normal eyelid position OS
- Conjunctiva/Sclera: white and quiet OU
- Cornea: clear OU
- Anterior Chamber: formed without hyphema or hypopyon OU
- Iris: flat and round OU
- Lens: WNL OU
- Dilated Fundus exam:
- Optic Nerve: sharp margins, no disc edema
- Macula: Healthy OU
- Vessels: Normal caliber OU
- Periphery: WNL OU
Vital signs and labs:
T: 36.8
HR: 104
RR: 24
BP: 112/60
- Sat: 99% RA
- CRP, BMP CBC: within normal limits
Imaging:

Image 1: Axial MRI T1 with contrast showing enhancement of the right 3rd nerve in the cisternal segment.

Image 2: Coronal MRI T1 with contrast showing enhancement and thickening of the cisternal portion of the right oculomotor nerve.
Differential:
- Post-Viral Neuritis
- Ophthalmoplegic migraine/Recurrent painful ophthalmoplegic neuropathy (OM/RPON)
- Aneurysm
- Schwannoma
- Tolosa-Hunt
- ADEM
- Miller-Fisher Syndrome
- TB, Lyme, Sarcoid, Lymphoma
Case Discussion:
Based on clinical presentation, the list of differential diagnoses for our patient’s third nerve palsy would be quite extensive. ADEM and Miller-Fischer syndrome can present as third nerve palsies but they would also tend to be accompanied by other neurologic deficits such as ataxia or areflexia. When the recurrent and resolving history is taken into account aneurysm, schwannoma, TB, Lyme, sarcoid, and lymphoma become less likely as these would tend to be slowly progressive and unlikely to resolve without treatment. Imaging confirms the absence of schwannoma and also rules out Tolosa-hunt which would show inflammation in the cavernous sinus and meningeal enhancement. Aneurysm, Tolosa-Hunt, and Miller-Fisher syndrome would also be quite rare in this age group.
With all of these factors taken together the most likely diagnosis is either post-viral neuritis or Ophthalmoplegic migraine/Recurrent painful ophthalmoplegic neuropathy (OM/RPON). It is possible that these two conditions are related.
Epidemiology
OM/RPON is a rare condition with a prevalence of 0.7 per million6; it usually begins in childhood.2,3 A systematic review authored by Gelfand found a median age of onset at 8 years old (interquartile range 3, 16). They also found that approximately 2/3 of patients affected by OM/RPON were female. 4
Signs and Symptoms
OM/RPON typically presents as a headache followed by ophthalmoplegia. If aheadache is present, it is typically unilateral and ipsilateral to the affected eye. The headache may or may not have migrainous features: photophobia, phonophobia, or nausea/vomiting. The ophthalmoplegia can occur immediately or up to 14 days after the headache. One or multiple nerves may be also be involved. The frequency of nerves affected in Gelfand’s paper was CN III 83%, CNVI 20%, and CNIV 2%. If multiple nerves were affected, CNIII was always included.4 When CNIII is affected it may be a partial or complete palsy. Both ptosis and mydriasis are common, especially among children.
Diagnostic Criteria
The diagnostic criteria have according to the International Classification of Headache Disorder 3-beta1 are:
- At least two attacks fulfilling criterion
- Unilateral headache accompanied by ipsilateral paresis of one, two or all three ocular motor
- Orbital, parasellar, or posterior fossa lesion has been excluded by appropriate
- Not better accounted for by another ICHD-3 diagnosis
As stated above, before the diagnosis of OM/RPON is made it is critically important to rule out other possible etiologies such as aneurysm, schwannoma, granulomatous disease, and inflammatory neuropathy.2
Pathophysiology
The pathophysiology of OM/RPON is a topic of ongoing debate. The diagnostic criteria in ICHD have been updated over the years to mirror the trends in the etiologic discussion by classifying it first as a migraine, then as a neuralgia, and most recently as a neuropathy.5 In fact, with the newest release of ICHD-3beta, the International Headache Society recommended the term ‘ophthalmoplegic migraine’ be abandoned.1 The driving force for this progression has been the need to explain the reversible contrast enhancement at the root entry zone of the affected cranial nerve.
Some who support the viewpoint of a neuropathy will point to the possibility of a benign neurotropic viral infection and highlight the similarities of the imaging in OM/RPON to those found in Bell’s Palsy.2 Others advocate for an immune-mediated neuropathy similar to chronic inflammatory demyelinating polyneuropathy (CIDP).5 In both of these explanations, the headache of OM/RPON is a result of inflammation of the nerve. The inflammation causes vasoconstriction of the nearby vasculature subsequently triggering the headache. However, these hypotheses are not without their weaknesses, CSF analysis and viral studies typically return unremarkable in OM/RPON.
Even with the current trend towards viewing OM/RPON as a neuropathy, there are still some who continue to advocate for a migrainous etiology. A paper published in 2014 proposed that a migraine induced vasospasm of the arteries supplying CNIII, IV, or VI could lead to ischemia of the nerves causing reversible breakdown of the nerve-blood barrier allowing for contrast extravasation and subsequent nerve enhancement on MRI.3 The authors make a great case, yet still leave questions unanswered. If migraine is the causal process then why does treatment with acute and prophylactic migraine medication offer little to no benefit or protection to patients?
In summary, there is still much work to be done towards reaching an understanding of how and why OM/RPON occurs. It is valuable to consider that OM/RPON may in fact have a multifactorial etiology resulting from processes and predispositions related to both neuropathy and migraine.
Treatment
With such ambiguity clouding the pathophysiology of OM/RPON it follows that there is also uncertainty regarding effective treatment for the condition.
Migraine medications both acute and prophylactic have been tried with unconvincing efficacy while the results of steroids have been positive yet mixed. Gelfand found steroids to be beneficial in 54% of cases reviewed. Yet the effects were either unclear, not beneficial or even harmful in 35%, 8%, and 4% respectively.4
Prognosis
Prognosis in OM/RPON is generally excellent and most patients can experience a full recovery in days to weeks. However small minority of patients may be left with persistent neurological deficits, especially those who suffer from repeated attacks.2,3,4,5
Faculty Approval by: Griffin Jardine, MD
Identifier: Moran_CORE_23858
References
- The International Classification of Headache Disorders, 3rd edition (beta version).
- Alexander, Al. Ophthalmoplegic Migraine: Reversible Enhancement and Thickening of the Cisternal Segment of the Oculomotor Nerve on Contrast-Enhanced MR Images. AJNR Am J Neuroradiol 19:1887–1891, November 1998
- Ambrosetto, Al. Ophthalmoplegic migraine: From questions to answers. Cephalalgia 2014, Vol. 34(11) 914–919
- Gelfand, Al. Ophthalmoplegic ‘‘Migraine’’ or Recurrent Ophthalmoplegic Cranial Neuropathy: New Cases and a Systematic Review. Journal of Child Neurology 27(6) 759-766. 2012
- Fö rderreuther, From Ophthalmoplegic Migraine to Cranial Neuropathy. Curr Pain Headache Rep (2015) 19: 21 DOI 10.1007/s11916-015-0492-1
- Quisling, Ophthalmoplegic Migraine: 30-year-old male with migraine headaches and occasional diplopia. http://webeye.ophth.uiowa.edu/eyeforum/cases/52-Ophthalmoplegic- Migraine-Diplopia-Headache.htm
Retinal Vasculitis
Home / Ophthalmic Pathology and Intraocular Tumors / Retina and Retinal Pigment Epithelium
Title: Retinal Vasculitis Case Report
Author: Sherief Raouf, Visiting Medical Student from the Stony Brook School of Medicine
Photographer: Glen Jenkins
Date: Friday, August 19, 2016
Image:


Diagnosis: Idiopathic mixed arterial/venous occlusive vasculitis
Secondary CORE Category: Retina and Vitreous / Focal and Diffuse Choroidal and Retinal Inflammation
Description of Image:
| Negative Laboratory Workup | |
| ANCA | Systemic necrotizing vasculitis |
| Serine Proteinase Ab | Systemic vasculitis |
| HbSAg, HbSAb | Hep B |
| ANA | Sensitive for SLE |
| Anti-Smith Ab | Specific for SLE |
| dsDNA Ab | Lupus Nephritis & SLE |
| Rh Factor | Rheumatoid Arthritis |
| Cardiolipin Ab and B2GP Ab | Anti-phospholipid syndrome |
| SSA Ab and SSB Ab | Sjogren’s & SLE |
| Ribonucleic Protein Ab | SLE & Mixed connective tissue disease |
| SCL-70 Ab | Scleroderma |
| PT, PTT, dRVVT | Hypercoagulability |
| Table 1. | |
Background: The patient is a 52 year old woman with a past medical history of diabetes mellitus and hypothyroidism who presented complaining of reduced vision OD, described as a “large grey spot in the center.” Visual acuity was 20/30 OD, 20/20 OS, pupils equal and reactive to light with no afferent pupillary defect, and the anterior chamber deep and quiet OU. The vitreous OD demonstrated +1 cells and the macula OD exhibited pigmentary atrophy and a small superior branch retinal artery occlusion (BRAO), while both eyes showed signs of periphlebitis.
There were no obvious signs of systemic inflammatory disease including Lupus, Sjogren’s, Giant-cell Arteritis, Granulomatosis with polyangiitis, or other systemic vasculitis. The review of systems was negative for cough, fever, orogenital ulcerations (Behçet disease), hearing loss and encephalopathy (Susac syndrome), or skin rash. Initial laboratory workup is shown in Table 1.
A laboratory investigation of infectious causes was performed and the following tests were negative: quantiferon, FTA-ABS, RPR, B. Burgdorferi Ab, toxoplasmosis and HIV ELISA. A laboratory workup for hypercoagulability was also negative (including PT, PTT, dRVVT). Brain MRI was negative for any acute intracranial process or findings of vascular inflammation or stenosis. Chest X-ray showed normal vascular markings, and a lack of any nodules, consolidations or hilar adenopathy.
The diagnosis of an arterial occlusive vasculitis was made, and treatment with oral Prednisolone and Mycophenolate mofetil was begun. One month later, the patient returned complaining of worsening blurriness OD. Exam revealed decreased vitreous cells with a persistent superior BRAO, periphlebitis and retinal neovascularization OD. The decision to treat the right eye with peripheral panretinal photocoagulation (PRP) was made. Over the ensuing months, the retinal vasculitis was medically managed with courses of prednisolone, mycophenolate and methotrexate and regular follow-up maintained. The photos above are taken from a visit 12 months after the initial visit, upon which a new BRAO was discovered.
The fundus photo demonstrates the gray-white sheathing of a retinal branch artery that is characteristic of a retinal vasculitis.1 The perivascular sheathing in occlusive vasculitis is thought to be an exudate of inflammatory cells around the vessel that leads to occlusion (Figure A, yellow arrow). Occlusion of the retinal vessel can result in ischemia of the retina and areas of capillary non-perfusion. This fundus photo demonstrates such an area of ischemic retina that is seen downstream and superior to the occluded vessel. This retina also exhibits neovascularization coincident with the ischemic areas of retina.
The late frame fluorescein angiography shows evidence of vascular obstruction in the inferior arcade, distal to the sheathed retinal branch artery. There is evidence of diffuse retinal vasculitis and vessel leakage. In addition, we can see the area of prior PRP of the areas of retinal capillary non-perfusion (Figure B, blue arrow). The angiography is able to additionally define the zones where the loss of retinal perfusion (Figure A, white arrow) has led to new areas of leaking neovascularization. Notably, there is a pronounced macular hyperfluorescence that corresponds to neovascularization (Figure B, green arrow). Given that neovascularization has continued, and that the vasculitis is active, the decision to restart corticosteroid treatment was undertaken with a plan for another round of PRP. Typically it would be preferable to ensure that the vasculitis is inactive when PRP is implemented, as its use can result in the release of more angiogenic factors, aggravating neovascularization.2
A key distinction to be made in making this diagnosis is between that of primary branch retinal vein occlusions, which closely mimics idiopathic retinal vasculitis. In BRVO, the occlusions typically occur at arteriovenous crossings, and are not multiple nor as peripheral as they tend to be in vasculitic obstructions.3 Finally, it should be said that there is some consensus to label idiopathic retinal vasculitis as Eales disease when vascular occlusions and neovascularization lead to recurrent vitreous hemorrhage in the presence of serological evidence of tuberculosis.3 Admittedly, this distinction may be a semantic one, as the yet unsolved pathophysiology may one day reveal these two entities to lie on one spectrum.
The differential for retinal vasculature occlusions is very broad and can be usefully divided into non-inflammatory (Diabetes, BRVO) and inflammatory causes. Inflammatory etiologies were examined with a careful history and an extensive laboratory and radiologic workup, yielding no clear cause. Indeed, many inflammatory retinal vascular obstructions are secondary to a systemic inflammatory process (infectious and non-infectious). However, primary idiopathic retinal vasculitis often is isolated to the eye and absent of any systemic involvement, frequently making this a diagnosis of exclusion.4
Format: Fundus photography and fluorescein angiography
References:
- Gass, J. Donald M. Stereoscopic Atlas of Macular Diseases: Diagnosis and Treatment. St. Louis: Mosby, 1997. Print.
- Biswas, J. et al. Eales disease–an update. Surv Ophthalmol 47, 197–214 (2002).
- Namperumalsamy, P., and Dhananjay S. “Eales ” Retina. Stephen Ryan MD et al. 5th ed. Oxford: Saunders, 2013. 1479-1485. Print.
- Saurabh, K., Das, R., Biswas, J. & Kumar, A. Profile of retinal vasculitis in a tertiary eye care center in Eastern India. Indian Journal of Ophthalmology 59, 297 (2011).
Faculty Approval By: Dr. Akbar Shakoor, Dr. Griffin Jardine
Identifier: Moran_CORE_23842
Disclosure: No financial disclosures to share