


Sickle Cell Retinopathy
Home / Retina and Vitreous / Other Retinal Vascular Diseases
Title: Sickle Cell Retinopathy
Author: Elizabeth Ann Urias, MSIV
Photographer:
Date: 06/24/2016
Image or video:

Keywords/Main Subjects: Sickle cell disease; sickle cell retinopathy; microvascular occlusion; retinal hemorrhage; salmon patch;
Diagnosis: Sickle Cell Retinopathy: Salmon Patch Hemorrhage
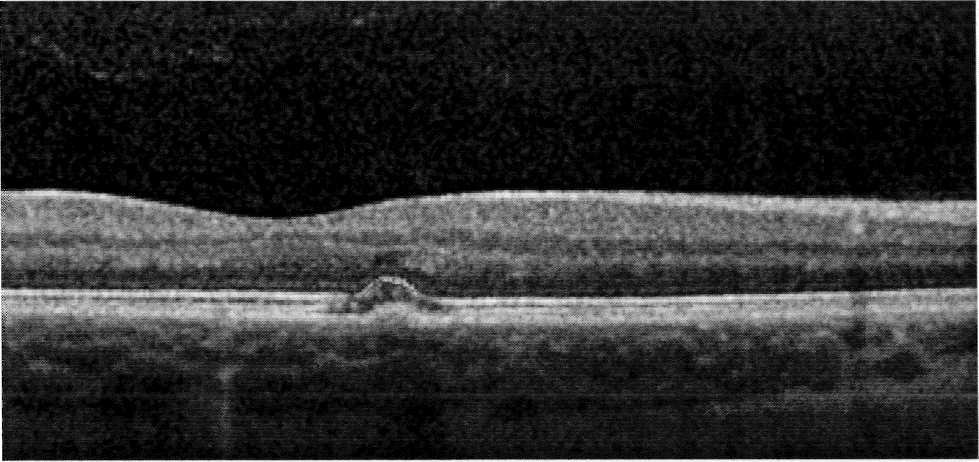
Description of Image: In sickle cell hemaglobinopathy, RBC sickling causes peripheral retinal arteriolar occlusion, leading to ischemic necrosis and weakening of the vessel walls. Fragile vessels result in hemorrhages in one or more layers of the retina. An intra-retinal hemorrhage is round or oval shaped, bright red, and measures ¼-1 disc diameter. In days or weeks, this bright red color becomes a salmon color, which is known as a “salmon patch.” After time, hemoglobin degradation occurs and the defect appears as bright yellow dots at several layers of the sensory retina, also known as “iridescent bodies.” (Bonanomi & Lavezzo, 2013)
References:
- Bonanomi, M. T. B. C., & Lavezzo, M. M. (2013). Sickle cell retinopathy: diagnosis and treatment. Arquivos Brasileiros de Oftalmologia, 76(5), 320–327.
Faculty Approval by: Griffin Jardine, MD
Identifier: Moran_CORE_21668
Financial Disclosures: None
Copyright statement: Copyright 2017. Please see terms of use page for more information.
Corneal Allograft Rejection Three Years Status Post Penetrating Keratoplasty
Home / External Disease and Cornea / Diagnosis and Management of Immune-Related Disorders of the External Eye
Title: Corneal Allograft Rejection Three Years Status Post Penetrating Keratoplasty
Author: Charlotte L. Marous, B.S., M.S.G.H.; Brian E. Zaugg M.D. July 2016
Photographer: James Gilman
Image or Video:

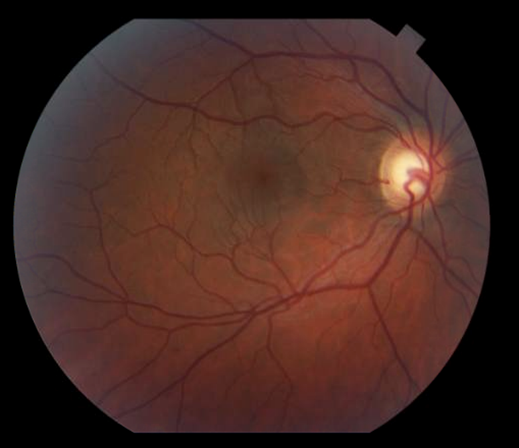
Figure 1: Slit lamp photo demonstrating healthy corneal graft with full clarity and intact sutures status post penetrating keratoplasty for keratoconus.
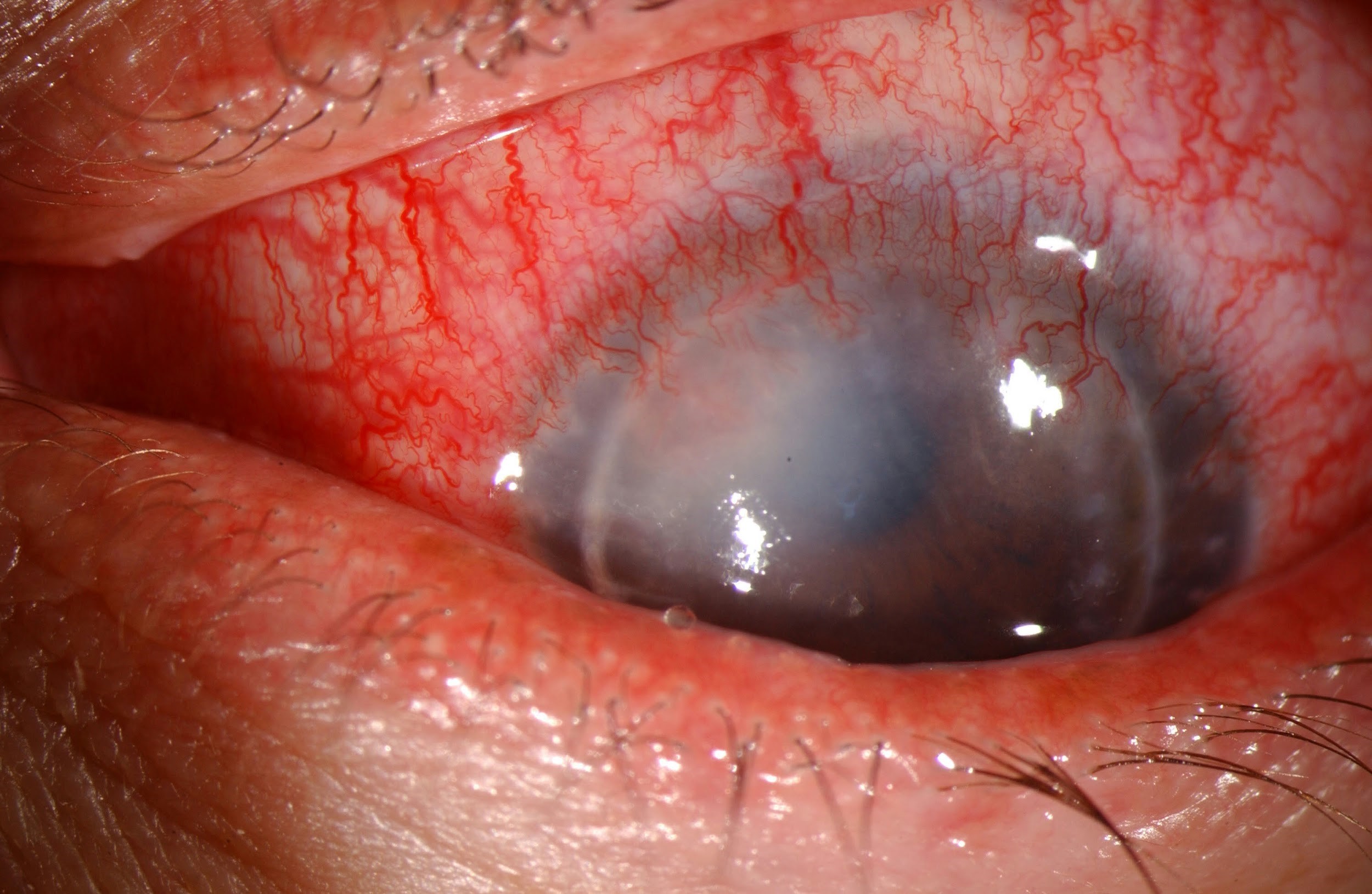
Figure 3 (right): Slit lamp photo of aggressive allograft rejection with active stromal vascularization and haze indicative of active graft rejection.

Figure 2 (left): Slit lamp photo of a penetrating keratoplasty with peripheral vascularization and stromal haze. The eye appears quiet and likely represents a chronic or past rejection.
Keywords/Main Subject: Keratoconus, Penetrating Keratoplasty, Transplant Rejection, Corneal Graft Rejection, Allograft Rejection, Corneal Vascularization, Immunosuppressives
Diagnosis: Corneal Transplant Rejection, Keratoconus
CASE
Chief Complaint: blurry vision of left eye for two weeks
History of Present Illness: A 35-year-old Caucasian female with bilateral keratoconus status post bilateral penetrating keratoplasty three years prior was referred for progressive blurry vision of the left eye for the past two weeks. Exam one year ago showed a healthy graft and clear cornea OU with visual acuity of 20/20 OD uncorrected and 20/20 OS corrected with a contact lens. The patient denied pain, discomfort, or discharge, stating only that the blurriness is “annoying.” No vision complaints OD. Using 1 drop of prednisolone acetate 1% once daily OU.
Past Ocular History: Keratoconus OU status post penetrating keratoplasty OU. Phacoemulsification with toric IOL for posterior subcapsular cataract OD, levator resection for ptosis OU, and a nuclear cataract OS currently stable under observation.
Medical history: FAP Gardner’s Syndrome
Family History: None
Social History: denies smoking, alcohol, illicit drugs
Medications: 1 drop prednisolone acetate 1% daily OU
Allergies: Morphine
Ocular Examination:
- Visual Acuity – best corrected with contact lens:
- OD – 20/25 +/-2
- OS – 20/200, 20/70 with pinhole
- Intraocular pressure (IOP by applanation):
- OD – 12 mmHg
- OS – 18 mmHg
- Pupils: No APD OU
- Slit lamp examinations:
- OD – cornea clear, penetrating keratoplasty, posterior chamber intraocular lens
- OS – cornea 3+ edema, superior rejection line, neovascularization 360 degrees, broken suture inferior
Assessment:
Corneal transplant rejection OS
Plan:
Prednisolone 1% every hour OS
Preservative free tears hourly OS
Follow-up in 2 weeks
CORNEAL ALLOGRAFT REJECTION DISCUSSION
Introduction:
Corneal graft transplant is the most widely practiced and successful type of solid organ transplantation in humans.1 Approximately 60,000 procedures are performed annually worldwide for diseases such as keratoconus, psuedophakic or phakic bullous keratopathy, trauma, infections, and corneal dystrophies or ectasias.1 It is associated with a high survival rate of 86% at 1-year post initial graft, largely attributed to the immune privilege of the eye.2 However, the 15-year graft acceptance rate declines to 55%, similar to survival rates in other forms of organ transplant.3 Graft rejection is one of the leading causes of corneal graft failure in the immediate and late postoperative period.1 Immune rejection and graft-related problems constitute the most important emergent presentations in graft patients. Long-term prophylactic use of topical steroids and immunosuppressive drugs can improve graft survival rate, but do not eliminate the risk, especially in “high-risk” recipients, and can be accompanied by side effects and potential toxicity.1 Novel biologic agents are showing promise in these patients unable to tolerate steroids.
Definition:
Corneal graft rejection is a reversible immune response against donor antigens. If signs of immunologic graft rejection do not clear within 2 months, the diagnosis of graft failure is made. Graft failure is the irreversible loss of graft clarity. It is important to rule out other common causes of loss of graft clarity before declaring failure. These include, but are not limited to infection, surgical trauma, glaucoma, and aging. Graft rejection is the most common cause of ultimate graft failure, accounting for >30% of cases.2
Pathophysiology:
The immune privilege of the cornea is maintained by the absence of blood vessels and lymphatics that deliver antigens to T cells in lymph nodes, scarcity of mature antigen presenting cells (APCs) in the central cornea, unusually low expression of major histocompatibility complex (MHC) antigens, and expression of the FAS ligand that induces apoptosis of stimulated Fas+ T cells.1 This privilege can be revoked by inflammation, infection, or trauma that induce neovascularization and lymphatic growth into the cornea; thus permitting APCs to enter the corneal stroma.1 Infection also induces pro-inflammatory cytokines that upregulate MHC antigen expression on corneal cells. Once the host immune system recognizes these foreign histocompatibility antigens on the cells of the corneal allograft, an immune cascade and response is initiated against these antigen, leading to eventual decompensation of the graft tissue.1 This rejection represents a form of delayed-type hypersensitivity response mediated by CD4+ T cells.2
Allograft rejection can affect one or more layers of the cornea (epithelium, stroma, and endothelium). The endothelium possesses minor regenerative properties and is a key player to maintaining corneal deturgescence.2 Thus, when >50% of the corneal endothelium is lost, graft rejection is likely to progress to graft failure.
Risk factors
Donor, host, and intraoperative factors influence graft rejection in corneal transplant recipients (Table 1). “High Risk” is defined as deep stromal vascularization of the host cornea of two or more quadrants, or a previous graft rejection in the affected eye.1
Table 1: Risk factors predisposing to graft rejection
| Donor
Factors |
· High antigenic load (depends on HLA/ABO compatibility between donor/host)
· Longer duration of storage of donor cornea (may reduce rejection) · Pretreatment of donor tissue with UV radiation (may reduce rejection by preventing activation of cytotoxic T cells) |
| Host
Factors |
· Vascularization of host cornea
· History of previously rejected graft (pre-sensitizes the host) · Ocular surface diseases (severe dry eye, chemical burns, radiation burns, ocular pemphigoid, Stevens-Johnson Syndrome, neuroparalytic disease) · Active keratitis · Pediatric patients (immune systemic of children is more active than adults)
|
| Intraoperative Factors | · Large/eccentric graft
· Synechiae at graft host junction · Penetrating grafts · Previous anterior segment surgical reconstruction · Bilateral graft · Suture removal (may trigger immune rejection) |
Clinical Signs and Symptoms:
Patients may be asymptomatic or may complain of increased blurry vision, redness, pain, irritation, or photophobia. Signs of graft rejection include:
- Stromal and/or epithelial edema
- Keratic precipitates (KPs) localized to the donor graft
- Corneal vascularization
- Stromal infiltrates
- Khodadoust line (separating immunologically damaged endothelium from unaffected endothelium)
- Elevated epithelial rejection line
- Krachmer spots (subepithelial infiltrates)
- Conjunctival injection
- Anterior chamber inflammation
Rejection can be classified based on corneal layer involvement (Table 2).1 Chronic focal endothelial rejection is the most common cause of graft rejection constituting 50% of cases. Epithelial rejection and stromal rejection represent roughly 2% and 1% of graft rejections, respectively.4
Table 2: Differentiating Features in Corneal Graft Rejection
| Epithelial | · Epithelial rejection line at the host graft junction
· Asymptomatic · No edema, keratic precipitates, or infiltrate |
| Stromal | · Circumferential limbal injection
· Patches of stromal infiltrate and haze · Stromal edema · Lymphocytes and plasma cells outside endothelial capillary wall |
| Endothelial | · Endothelial line
· Stromal and epithelial edema · Keratic precipitates on graft endothelium · Cell and flare possible, but difficult to visualize due to edematous cornea |
Adapted from Panda A, Vanathi M, Kumar A, et al. Corneal graft rejection. Survey of Ophthalmology. 2007; 52: 375-95.
Differential Diagnosis of Corneal Graft Rejection:
- Suture abscess or corneal infection
- Uveitis
- Epithelial downgrowth
- Recurrent herpetic keratitis
- Sterile or infectious endophthalmitis
- Increased IOP
Workup
Diagnosis is based on history and slit lamp examination. Important questions to be considered include: time since corneal transplantation, current eye medications, compliance with eye medications and postoperative follow-up, recent changes in topical steroid regimen, and inquiry into the initial indication for the corneal transplant. On slit lamp exam, inspect for endothelial rejection line, keratic precipitates, subepithelial infiltrates, edema, and cells in anterior chamber.
Prevention Practices and Immunosuppressive Agents:
Prevention can be subdivided into preoperative, intraoperative, and postoperative measures. Preoperatively, minimizing the antigenic difference between the host and donor tissue (tissue matching) and reducing the antigenic load of the donor tissue (UV light exposure) can help diminish the risk of rejection. Intraoperative measures of preventing immune-mediated allograft rejection are achieved by meticulous surgical techniques including good graft-host apposition and sterile suturing. Close follow-up monitoring for IOP, intact sutures, and compliance to immunosuppressive medications contribute to post-operative risk reduction.
Immunosuppressive medications play a fundamental role in the reduction of graft rejection. Current practice guidelines suggest “low risk” patients be put on long-term once daily prophylactic topical steroids (1% prednisolone drops), while “high risk” recipients adhere to a more stringent prophylactic regimen of long-term daily topical steroid plus a systemic immunosuppressive.1 Topical and oral steroids control the host immune system by preventing invasion by IL-1 and IL-6 producing macrophages and initiation of T-cell responses. Given the side effect profile of prolonged steroid use including cataract formation, glaucoma, and impaired wound healing, newer preventative therapeutics are becoming more widely available. Calcineurin inhibitors including cyclosporine (CsA), tacrolimus (FF-506), and mycophenolate mofetil (MMF) have been shown to be effectively substituted for steroids.5 Intraocular delivery of immunosuppressants in “high risk” rabbits was associated with reduced rate of graft rejection compared to those treated with topical or oral agents alone; supporting another potential route of prophylaxis.2 Novel biologic agents blocking receptors of TNF-a, VEGF, and CCL2 are also showing promise as prophylactic treatment in patients affected by high immune risk and corneal neovascularization.6 These agents work by reducing corneal inflammation, vascularization, angiogenesis, lymphangiogenesis; all factors common to graft rejection.2
Treatment
Despite prophylactic efforts, graft rejection may transpire. Management is aimed at reducing the active immune response. The likelihood of reversibility is largely dependent on the corneal layer affected. For epithelial and subepithelial acute rejections, which have a higher rate of reversibility, primary treatment involves topical steroids (1% prednisolone) six times per day along with preservative-free artificial tears that hydrate the ocular surface.7 Severe endothelial rejection requires hourly topical steroids in combination with systemic therapy (40 to 80 milligrams of oral prednisone daily, or 1-2 intravenous doses of 400 milligrams of methylprednisolone with or without subconjunctival betamethasone 3 milligrams in 0.5 milliliters).7 Careful monitoring for IOP and resolution of the inflammation should be assessed every 3-7 days until improvement is noted, at which point steroids may be slowly tapered and maintained at a long-term low dose.
Alternatives to Normal Corneal Tissue Grafts in “High Risk” Patients:
One alternative for patients with multiple failed corneal grafts is a Boston keratoprosthesis, an artificial cornea. This is a synthetically generated cornea FDA-approved for use in patients with severe corneal opacification and high risk of transplant failure. Donor corneal tissue is secured between a rigid polymethyl methacrylate optic front plate and back plate lined with holes to allow for communication with the aqueous for nutrition and hydration of the cornea. The plates are snapped together and sutured into the recipient eye, similar to a typical corneal transplant.8-9 A prospective case series of 141 cases across 17 centers reported visual acuity of 20/40 in 23% of patients and 20/200 in 57% of patients after keratoprosthesis placement.10 Failure for visual acuity improvement and post-operative complications included epithelial defects, stromal thinning, dellen formation, retroprosthetic membrane formation, retinal detachment, and development of glaucoma.2,11
Bioengineered corneal equivalents represent another possible option. These biosynthetic implants are equivalent to corneal stromal extracellular matrix and are based on chemically crosslinked collagen designed to function as regeneration templates. The idea is that regeneration of endogenous corneal layers and functional corneal nerves occur in the collagen matrix, thus hydrogel implants that mirror this natural cornea structurally may promote active regeneration of endogenous corneal epithelial and stromal cells.2 A recent 4-year follow-up clinical study provided strong evidence supporting high acceptance and adaptation of the hydrogel with improved visual acuity and central nerve ingrowth.12 The major limitation of this method resides in the fact that positive outcomes are limited to lamellar keratoplasty, in which host endothelium is still intact. This would not be helpful in patients requiring penetrating keratoplasty.
References:
- Panda A, Vanathi M, Kumar A, et al. Corneal graft rejection. Surv Ophthalmol. 2007;52:375-95.
- Yu T, Rajendran V, Griffith M, Forrester JV, Kuffova L. High-risk corneal allografts: a therapeutic challenge. World J Transplant. 2016;6:10-27.
- Williams KA, Esterman AJ, Bartlett C, Holland H, Hornsby NB, Coster DJ. How effective is penetrating corneal transplant? Factors influencing long-term outcome in multivariate analysis. Transplantation. 2006;81:896-901.
- Guilbert E, Bullet J, Sandali O, Basli E, Laroche L, Borderie VM. Long-term rejection incidence and reversibility after penetrating and lamellar keratoplasty. Am J Ophthalmol. 2013;155:560-69.
- Hill JC. Systemic cyclosporine in high-risk keratoplasty: long-term results. Eye (Lond). 1996;9:422-28.
- Fasciani R, Mosca L, Giannico MI, Ambrogio SA, Balestrazzi E. Subconjunctival and/or intrastromal bevacizumab injections as preconditioning therapy to promote corneal graft survival. Int Ophthalmol. 2015;35:221-7.
- Bagheri N, Wajda BN. The Wills Eye Manual: office and emergency room diagnosis and treatment of eye disease, 7th edition. Philadelphia: Lippincott Williams and Wilkin, 2017:98-99.Print.
- Ilhan-Sarac O, Akpek EK. Current concepts and techniques in keratoprosthesis. Curr Opin Ophthalmol. 2005;16:246-50.
- Birkholz ES, Goins KM. Bostom keratoprosthesis: an option for patients with multiple failed corneal graft. March 9, 2009; Available from: http://www.EyeRounds.org/cases/94-Boston-Keratoprosthesis-Failed-Corneal-Graft.htm
- Zerbe BL, Belin MW, Ciolino JB. Boston type 1 keratoprosthesis study group, results from the multicenter Boston Type 1 Keratoprosthesis study. Ophthalmol. 2006;113:1779.
- Ray S, Khan BF, Dohlman CH, D’Amico DJ. Management of vitreoretinal complications in eyes with permanent keratoprosthesis. Arch Ophthalmol. 2002; 120: 559–566.
- Fagerholm P, Lagali NS, Ong JA, Merrett K, Jackson WB, Polarek JW, Suuronen EJ, Liu Y, Brunette I, Griffith M. Stable corneal regeneration four years after implantation of a cell-free recombinant human collagen scaffold. Biomaterials. 2014; 35: 2420-27.
Faculty Approval by: Brian E. Zaugg, M.D.
Copyright: Charlotte L Marous © 2016. For further information regarding rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Identifier: Moran_CORE_21656
Disclosure (Financial or other): The authors have no financial conflicts of interest.
UNDER REVIEW
Familial Exudative Vitreoretinopathy
Home / Pediatric Ophthalmology and Strabismus / Disorders of the Retina and Vitreous
Title: Familial Exudative Vitreoretinopathy
Author (s): Jeffrey Z. Kartchner, BS; Mary E. Hartnett, MD
Photographer: Unknown
Date: 07/20/2016
Keywords/Main Subjects: Familial exudative vitreoretinopathy, exudative retinal detachment, persistent fetal vasculature, retinopathy, leukocoria
Secondary CORE Category: Retina and Vitreous / Diseases of the Vitreous and Vitreoretinal Interface
Diagnosis: Familial Exudative Vitreoretinopathy
Format: Case presentation
History of Present Illness: A 4-month old boy born at full term after an uncomplicated pregnancy presented with exophoria and microphthalmia of the left eye, and also with right eye preference. He was seen by an outside pediatric ophthalmologist and diagnosed with leukocoria and probable persistent fetal vasculature (PFV) and referred to our retina service for surgery.
Past medical history was unremarkable. Family history was positive for mother with strabismus and maternal uncle with congenital cataract of unknown etiology.
Initial Examination: The patient’s visual acuity was “fix & follow” in both eyes. He had a mild afferent pupillary defect in the left eye, but none present in the right eye. His corneal diameter on the left side was 10mm, compared to 11mm on the right. External exam was otherwise normal bilaterally. Fundus exam of the right eye was normal, but no view was achieved on the left eye because of a dense retrolental membrane. A B-Scan ultrasound was performed, which showed a total retinal detachment with an open funnel. The decision was made to proceed with examination under anesthesia (EUA) with plans to perform a lensectomy and vitrectomy in the left eye for presumed PFV.
Clinical Course: The patient underwent EUA and was found to have an area of peripheral avascular retina in the right eye. Fluorescein angiography of this eye later showed peripheral avascular retina and leakage of the vessels at the junction of vascular and avascular retina consistent with stage 2A FEVR (Figure 1). The patient underwent lensectomy and vitrectomy in the left eye which revealed a retrolental membrane and a complete retinal detachment with a nasal exudative retinal fold, consistent with Stage 5 FEVR (Figure 2).
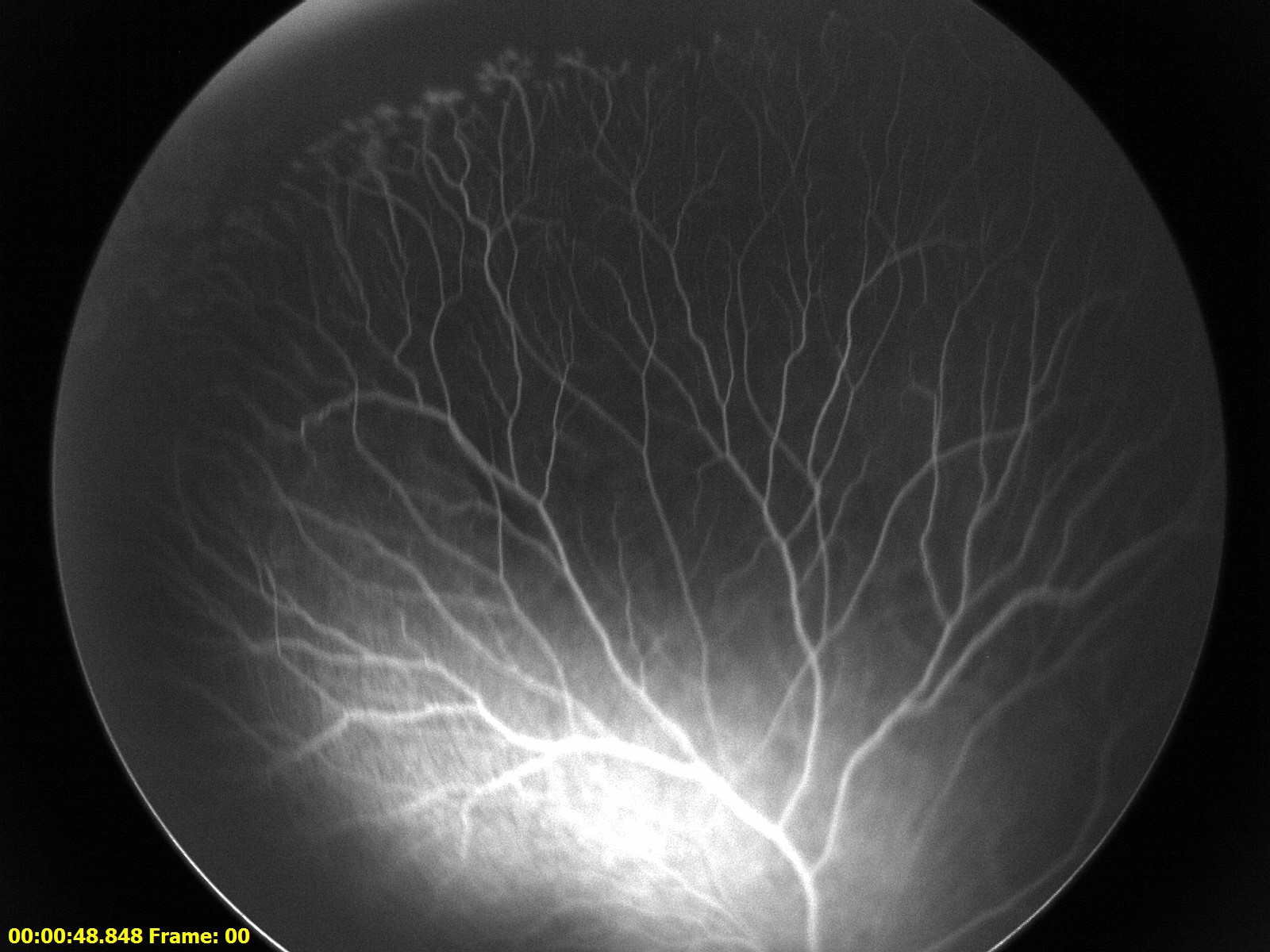
Figure 1: Fluorescein angiography (FA) of the right eye. There is an area of peripheral avascular retina with neovascular tufts along the border.
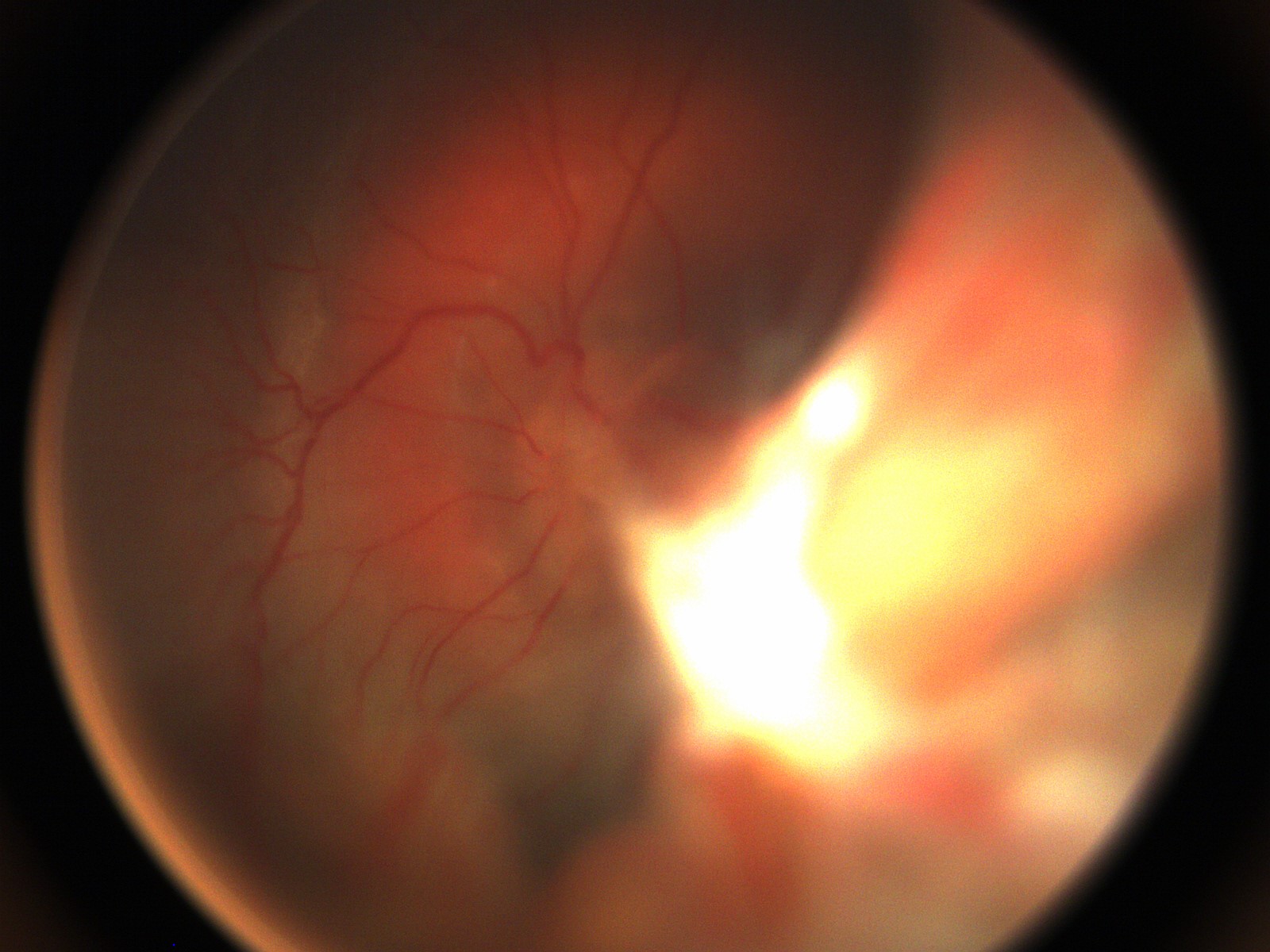
Figure 2: Fundus photography of the right eye following initial lensectomy-vitrecomy, demonstrating the nasal fold from the surgeon’s view and a circumferential subretinal band surrounding the optic nerve.
The patient underwent scatter photocoagulation of the avascular retina in the right eye (Figure 3). The patient subsequently underwent several additional EUAs and surgeries to drain subretinal fluid or release traction. Genetic studies revealed a heterozygous mutation of the FZD4 gene via the EyeGene research study.

Figure 3: FA of the right eye. Scatter photocoagulation has been performed to include the avascular retina and neovascularization in its entirety.
Current Exam: At age 21-months the patient’s visual acuity in the right eye is 20/32 by Allen acuity cards and light reactive in the left eye. There is a 3+ afferent pupillary defect in the left eye. Disease in the right eye has been stable after scatter photocoagulation. The left eye shows reattachment of the retinal detachment with resolving subretinal exudates and the persistent subretinal band and nasal fold (Figure 4). Current management includes regular monitoring and maximizing his visual outcome with consideration of the aphakic correction of the left eye.

Figure 4: Fundus photography of the left eye. Intra-retinal cholesterol crystals remain after some resolution of exudates. A sub-retinal band with persistent exudates is observed extending anteriorly.
Discussion:
FEVR is a genetically inherited disease with multiple modes of inheritance, including autosomal dominant, autosomal recessive, and X-linked. Expressivity is variable, such that it is common for patients to present with widely different phenotypes, even between eyes in the same individual. Up to 58% of relatives of patients diagnosed with FEVR have asymptomatic clinical findings1. Mutations in FZD4, LRP5, NDP and TSPAN12, members of the Wnt signaling pathway, are implicated in about 50% of cases. Newly discovered mutations in ZNF408 have also been shown to lead to disease2.
Genetic mutations lead to incomplete vascularization of the peripheral retina in full-term infants that may progress to neovascularization. The disease appears similar to acute retinopathy of prematurity (ROP). Exudative retinal detachment, retinal folds, macula ectopia, fibrous bands, and tractional retinal detachments also occur with FEVR progression. Studies have been performed evaluating the location of retinal folds in children with FEVR3,4. It was shown that patients with retinal folds extending from the posterior pole to the lens present in such a way that the diagnosis of FEVR can be difficult to distinguish from PFV, as was the case in the current patient presentation. It is, therefore, imperative to perform EUA with careful examination of the contralateral eye for any signs of FEVR.
Management of FEVR includes early screening in suspected cases, with prompt laser photocoagulation therapy or cryotherapy to any avascular retina and neovascularization. Retinal detachments are managed surgically, with frequent follow-up for monitoring of repeat detachments. Recent studies have been performed evaluating the efficacy of anti-VEGF agents in the treatment of FEVR. While initial results can be promising, the long-term therapy in these patients did not provide sustained positive outcomes5. Limitations to research regarding FEVR include the small patient population and wide variability of expressivity between patients.
References:
1. Trese MD et al. High prevalence of peripheral retinal vascular anomalies in family members of patients with familial exudative vitreoretinopathy. Ophthalmology. 2014;121(1):262-8.
2. Collin RW, Nikopoulos K, Dona M, Gilissen C, Hoischen A, Boonstra FN et al. ZNF408 is mutated in familial exudative vitreoretinopathy and is crucial for the development of zebrafish retinal vasculature. Proc Natl Acad Sci USA. 2013; 110: 9856–9861.
3. Robitaille JM et al. Phenotypic Overlap of Familial Exudative Vitreoretinopathy (FEVR) with Persistent Fetal Vasculature (PFV) Caused by FZD4 Mutations in two Distinct Pedigrees. Ophthalmic Genet. 2009;30(1):23-30
4. Trese MD et al. Clinical Presentation of Familial Exudative Vitreoretinopathy. Ophthalmology. 2011;118(4):2070–2075.
5. Henry CR. Long-term follow-up of intravitreal bevacizumab for the treatment of pediatric retinal and choroidal diseases. J AAPOS. 2015 Dec;19(6):541-8.
Faculty Approval by: Mary E. Hartnett, MD
Copyright statement: Copyright ©2015. For further information regarding the rights to this collection, please visit: Terms of Use to copyright information page on Moran CORE
Identifier: Moran_CORE_21639
Disclosure (Financial or other): None
UNDER REVIEW
Aniridia
Title: Aniridia
Authors: Judd Cahoon, PhD; Dr. Jeff Pettey, MD
Photographer: James Gillman, CRA, FOPS, Project Administrator Ophthalmic Imaging Date: 7/21/2015
Keywords: Aniridia
Diagnosis/Differential Diagnosis: Aniridia, salzmann’s nodules, non-proliferative diabetic retinopathy
Description of Case:
42-year-old male with congenital aniridia presents for annual diabetic screen. He has multiple medical comorbidities including type-1 diabetes since the age of 12 and congenital partial agenesis of the corpus callosum. His diabetes is poorly controlled (A1C of 8). The patient states his vision (both near and far) is worsening over the past few years. He states he bumps into things often because he doesn’t see them and this has led to some falls.
Past Ocular History: Cataract surgery in both eyes in 2011
Past Medical and Surgical History: T1DM since age of 12, no other surgeries Medications: Novolin subq daily.
Family History: Mother and father with history of diabetes mellitus, no history of aniridia in family.
Social History: Lives in a house with others who help him make his food. He smokes 8-10 cigarettes a day. No alcohol use or drug use.
Ocular Exam:
• External exam: Normal both eyes (OU)
• Distance visual acuity (Snellen)
o Right eye (OD) 20/200, no improvement with pinhole
o Left eye (OS) 20/250, no improvement with pinhole
• Pupils
o OD: Dark 7mm, light 7 mm, no reaction
o OS: Dark 7mm, light 7mm, no reaction
• Motility: Full OU, nystagmus present in all directions
• Visual Fields
o OD: all peripheral fields decreased
o OS: decreased in inferior nasal and temporal fields
• Tonometry (Applanation)
o OD: 16 mmHg
o OS: 16 mmHg
• Anterior Segment exam (Figure 1)
o Lids and lashes: Normal OU
o Conjunctiva and Sclera: white and quiet OU
o Cornea: Inferior circular subepithelial opacity – likely Salzmann’s nodules, OU
o Anterior Chamber: Deep and quiet OU
o Iris: Aniridia OU
o Lens: Posterior chamber intraocular lens, open posterior capsule (OD), PCIOL with 1+ poster capsular opacification (OS)
o Vitreous: normal OU
• Fundus Exam:
o Disc: atrophic area inferior to nerve (OD), normal (OS)
o C/D ratio: 0.6 (OD), 0.65 (OS)
o Macula: normal OU
o Vessels: normal OU
o Periphery: inferonasal circular retinal scar next to optic nerve (OD), normal (OS)
Discussion
Aniridia, or absence of the iris, is estimated to have a prevalence of about 1.8 per 100,000 in the population. It is inherited commonly in an autosomal dominant fashion with complete penetrance but variable expressivity (Familial congenital aniridia), but can also be acquired in both sporadic form (Miller syndrome) or, rarely, in an autosomal recessive fashion (Gillespie Syndrome). Additionally, aniridia can be acquired after trauma. Caused by mutations a gene on chromosome 11p13, PAX6 is involved in regulating transcription in the cornea, lens, ciliary body, and retina, genetic testing can be acquired in patients presenting with aniridia.
Symptoms
Patients with aniridia often present with bilateral absence of the iris, which can be either complete or partial. Caregivers can often notice severe light sensitivity, or photophobia, in the child. Multiple ocular comorbidities are also noted including dry eye, meibomian gland dysfunction, and (as seen in our patient) nystagmus, decreased visual acuity, corneal opacification, aniridia-associated keratopathy, cataracts, and optic nerve hypoplasia. Glaucoma is also of major concern and often develops due to inhibition of aqueous flow by the iris stump blocking the trabecular meshwork. Tonometry, pachymetry, and optic nerve exams, and visual field testing are all useful in the diagnosis of an individual with suspected aniridia. However, aniridia remains a clinical diagnosis that can be supported by genetic testing (for PAX6 mutations). A careful family history is also required to rule out other syndromes.
Clinicians should be suspicious of syndromic sequelae in individuals with sporadic aniridia. WAGR syndrome (Wilms tumor, aniridia, genitourinary abnormalities, and mental retardation) is caused by deletion of the short arm of chromosome 11, which, in addition to PAX6, houses the WT1 gene. Presentation of WAGR syndrome includes characteristic facies (long face, stubby nose, long philtrum) in addition to renal and genital abnormalities. Bardet-Biedl Syndrome is an autosomal recessive disease which, like WAGR syndrome, causes kidney and genitourinary abnormalities, but also includes retinal pigment atrophy, zonular cataracts, and polydactyly.
Treatment
Treatment for aniridia can include colored contact lenses with dark periphery or filter/tinted lenses of photophobia. Importantly, treatment of ocular co-morbidities is paramount.
Preservative free tears may be used to for associated keratopathy and dry eye syndrome. Limbal stem cell transplants with amniotic membranes can prevent corneal pannus and scarring. Regular dilated eye exams are useful to follow optic nerve changes along with pressure checks and visual fields. If genetic testing was not obtained, regular abdominal ultrasounds are useful to screen patients for Wilms tumor.
Images:

Figure 1: Traumatic aniridia of the right eye.

Figure 2: Congenital aniridia, left eye. Initial examination shows a large, dark, and dilated pupil that does not react to light.

Figure 3. Aniridia can also present as partial absence of the iris as demonstrated here, with residual iris on the inferior margin. Of note, there is almost always residual iris in all cases of aniridia, but the iris might be so small as only to be visible with careful exam or gonioscopy.
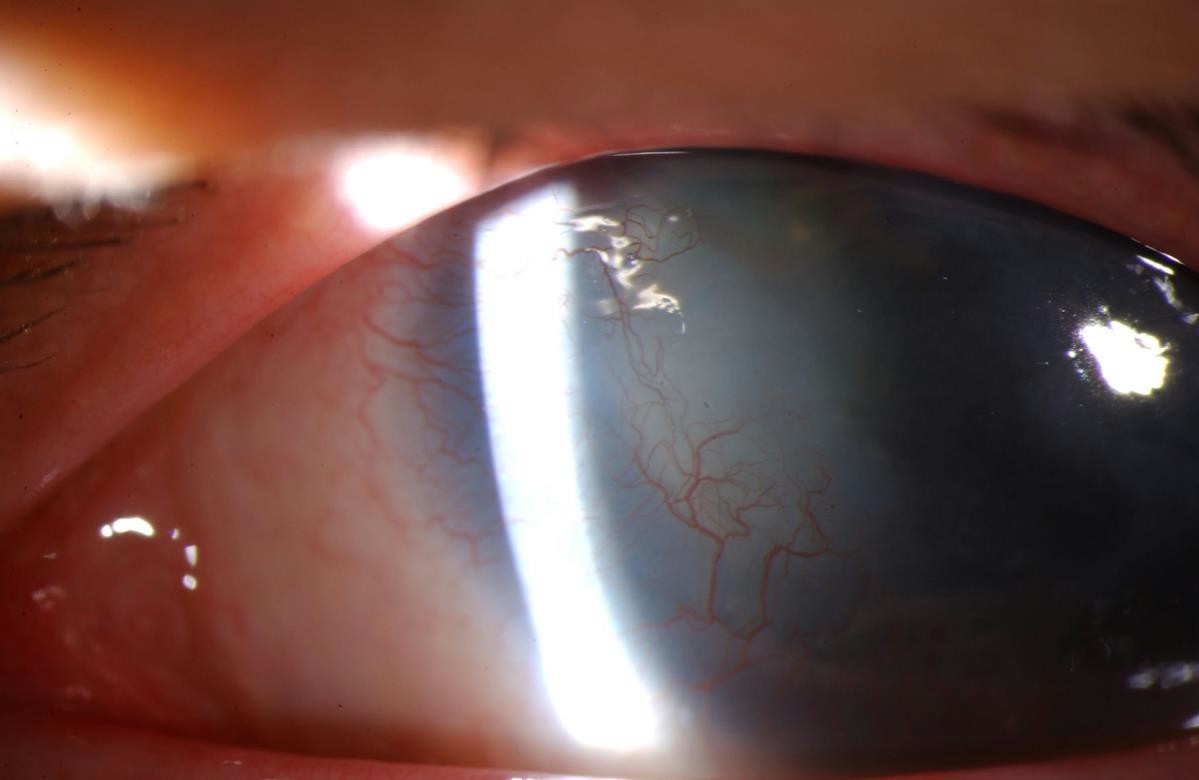
Figure 4. Aniridia with corneal pannus and associated neovascularization.
Summary of Case:
Vision usually remains poor in patients with aniridia, around 20/200, thus preserving the vision patients have is of utmost importance. In our patient, the nystagmus and corneal disease were not uncommon findings for someone with aniridia. Neither were his visual field deficits, though his pressure was normal. He had his cataracts removed previously and will be referred to a corneal specialist to follow his Salzmann’s nodules. The patient previously underwent YAG laser for PCO and has some remnant opacifications that could benefit from further treatment. Fortunately, his diabetes has not affected his eyes clinically, yet.
References:
- Goetz K, Vislisel JM, Raecker ME, Goins KM. Congenital Aniridia. March 10, 2015; Available from: http://EyeRounds.org/cases/211-Aniridia.htm
- Hingorani, M., Hanson, I., Van Heyningen, V. Aniridia. European Journal of Human Genetics 2013; 20:1011-1017.
- Kokotas, H. et al: Clinical and molecular aspects of aniridia. Clin Genet. 2010, 77(5):409-20.
- Lee, H., Khan, R. and O’Keefe, M. Aniridia: current pathology and management. Acta Ophthalmologica 2008; 86:708–715.
Copyright statement: Copyright Judd Cahoon, 2016. Please see terms of use page for more information.
UNDER REVIEW
Punctate Inner Choroidopathy (PIC)
Home / Retina and Vitreous / Other Retinal Vascular Diseases
Title: Punctate Inner Choroidopathy (PIC)
Author: Sai Bhuvanagiri (MS IV); Akbar Shakoor, MD
Photographer: Photo courtesy of Dr. Shakoor
Date: 09/14/2015
Financial Disclosure: None
Image: SD-OCT of images of Active Punctate Inner Choroidopathy (above) with Choroid Neovascular Membrane complication and Inactive Punctate Inner Choroidopathy lesion (below)

Keyword/ Main Subject: SD-OCT images of Active Punctate Inner Choroidopathy (PIC) with complication of Choroidal Neovascular Membrane (CNV) (above) and bottom image of lnactive Punctate Inner Choroidapthy of the same patient after appropriate anti-VEGF treatment
Diagnosis/ Differential Diagnosis: Punctate Inner Choroidopathy, Multiple Evanescent White Dot Syndrome, Multiple Choroiditis, Presumed Ocular Histoplasmosis Syndrome, Birdshot’s Retinopathy
Brief Description: Punctate Inner Choroidopathy (PIC) is a very rare ocular inflammatory disease considered to be part of the White Dot syndromes that fall under the umbrella of uveitis. PIC frequently affects young Caucasian myopic women with mostly bilateral involvement 2.
Patients present with symptoms of loss of central visual acuity (VA), photopsia and central scotoma. PIC is usually a benign disease; however, visual loss can occur secondary to complications of CNV or subretinal fibrosis. The current etiology is unknown but is thought to have familiar predisposition to autoimmune/ inflammatory diseases with response against antigens in the outer retina and inner choroid and increase in choroidal thickness during acute phases of PIC and reduction during regression of PIC 4 • The mainstay of treatment for PIC is anti-VEGF intravitreal injections with mean visual acuity (VA) that returned from baseline of
3.2 /10 to 6/10 in 12 months with no disease recurrence 6. Other treatment option includes oral steroids with VA return from baseline of 1/10 to 6.7/10 in a week 5. However, they are not the primary form of treatment due to systemic side effects. Other treatment options include mycophenolate mofetil, laser photocoagulation, and submacular surgery 1.
When this patient was diagnosed with PIC, she initially presented with central vision loss in the right due to CNVM. On further investigation of patient’s history and ruling out infectious causes, the patient was found to have PIC. The main factors that lead to the diagnosis of PIC are patient’s acute symptoms, patient’s demographic correlation to similar patients with PIC, lack of anterior chamber inflammation and SD-OCT image (above) finding of RPE elevation with a loss of photoreceptor (PR) layer 3. The patient was treated with Avastin (anti-VEGF) intravitreal injections with week duration to treat both CNVM and PIC. The patient is followed every three months and had no recurrences so far for the past nine months. The latest SD-OCT image confirms the resolution of the patient’s active PIC phase with re-appearance of her PR layer and reduced RPE layer elevation 2.
Identifier: Moran_CORE_ 21453
References
- Campos J, et al. “Punctate Inner Choroiditis” Innovations in Ophthalmology. October, 2014.
- Channa R, et al. “Characterization of macular lesions in punctate inner choroidopathy with spectral domain optical coherence tomography “Journal of Ophthalmic Inflammations and Infections. July, 2012.
- Goldstein D, et al. “Multifocal Choroiditis vs. PIC: Variations on a Theme”.
Ophthalmology Review, July, 2004. - Hiraoka K, et al. “Increased macular choroidal blood flow velocity and decreased choroidal thickness with regression of punctate inner choroidopathy “BMC Ophthalmology. May, 2014.
- Olsen TW, Capone A, Sternberg P, et al. Subfoveal choroidal neovascularization in punctate inner choroidopathy. Surgical management and pathologic findings. Ophthalmology. 1996.
- Zhang X, Wen F, Zuo C, Li M, Chen H, Huang S, Luo G. Clinical features of punctate inner choroidopathy in Chinese patients. Retina.
Copyright statement: Sai Bhuvanagiri, ©2015. For further information regarding the rights to this collection, Please see terms of use page.
Central Serous Chorioretinopathy – Case Report
Home / Retina and Vitreous / Choroidal Disease
Title: Central Serous Chorioretinopathy
Author: Sravanthi Vegunta, Eileen S. Hwang, Mary E. Hartnett
Photographer: Mel Chandler
Date: 08/05/2015
Keywords/Main Subjects: Central serous chorioretinopathy; Scotoma; Metamorphopsia; Retinopathy; Corticosteroid use; Smoke stack
Diagnosis: Central serous chorioretinopathy
Brief Description: History of Present Illness: The patient was a 46 year old female with a history of hypothyroidism, depression and seasonal allergies who presented 5 weeks after the sudden onset of a floaters, a paracentral scotoma and distortion in her left eye. At first, her central vision was primarily affected, but shortly before her presentation in our clinic, the affected area spread to include an area to the right as (Figure 1). She denied visual problems with her right eye.
Her medical history was significant for the use of intranasal steroid spray for seasonal allergies. She had stopped this a few weeks prior to presentation after a visit to an outside ophthalmologist.

Figure 1: Amsler grid test of the left eye revealed a central grey area and a superior area of distortion.
Initial examination: The patient’s visual acuity with correction was 20/20 in the right eye and 20/60 in the left eye. Intraocular pressure was 16 mmHg in the right eye and 16 mmHg in the left eye. She did not have a relative afferent pupillary defect. Confrontation visual fields and extraocular motility were intact. Anterior segment slit lamp examination was normal in both eyes. On fundus examination, both optic nerves were normal in appearance with a cup-to-disc ratio of 0.3 in the right eye and 0.2 in the left eye. The macula of her right eye was normal with a good foveal reflex. Her left macula had an area of pigment near the fovea (Figure 2) and the retina appeared elevated in the central macula, consistent with subretinal fluid (Figure 3). The retinal vasculature and periphery were normal in both eyes.

Figure 2: Color fundus photography. A) The right macula and fovea appear normal. B) In the left eye, the foveal reflex appears yellow and blunted. There are parafoveal pigmentary changes. There is an area of retinal elevation consistent with subretinal fluid in the central retina.
Clinical Course: Patient underwent optical coherence tomography (OCT; Figures 3 and 4) and fluorescein angiography (FA; Figure 6) of each eye.

Figure 3: Left eye OCT demonstrating subretinal fluid in the central macula.
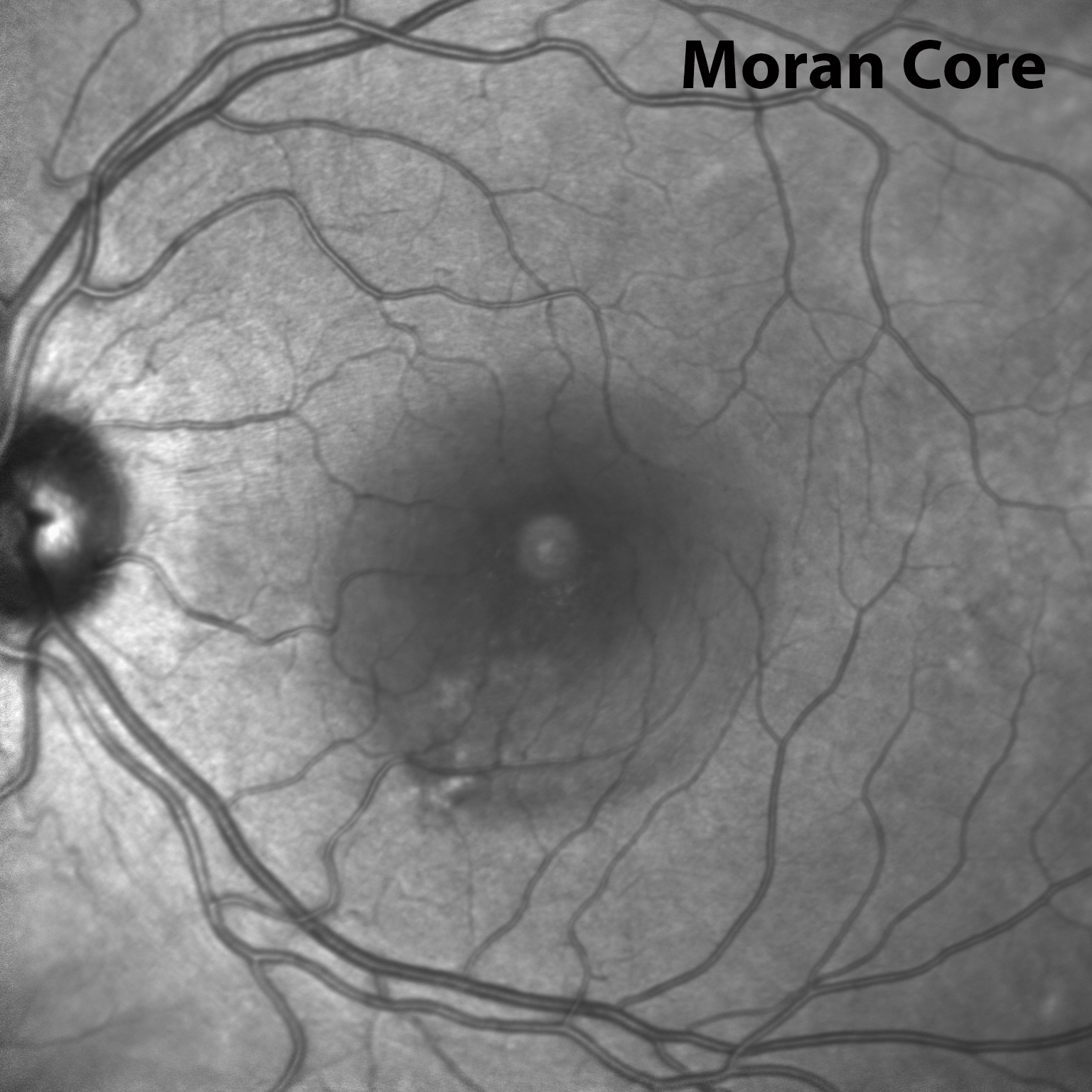
Figure 4: Left eye infrared image showing a hyporeflective area in the central macula corresponding to the area of subretinal fluid.

Figure 5: Fluorescein angiography of the left eye. There are window defects and late phase leakage from a point in the inferior macula toward the fovea in a smoke stack configuration.
The patient was diagnosed with central serous chorioretinopathy (CSCR) and informed that her visual changes were likely to improve without intervention. Information was provided to the patient regarding the limited quality data for treatment of CSCR with spironolactone, rifampin, epleronone, and low dose aspirin. Low dose aspirin was recommended to the patient since there was a controlled study supporting its use, and the risks were felt to be relatively benign.
Discussion:
CSCR is characterized by a build up of subretinal fluid in the macula caused by abnormalities of the choroidal circulation.1 Fluid leaks from the choroidal circulation and passes through hyperpermeable areas of the retinal pigment epithelium (RPE), accumulating in the subretinal space. 2
While the specific cause of CSCR is not well understood, some systemic risk factors have been associated with CSCR. In particular, corticosteroid administration and stressful events (presumably associated with high endogenous corticosteroid levels) are associated with the development of CSCR.2 The patient described in this case report was using intranasal steroids, which have specifically been associated with CSCR in addition to systemic, intraarticular, and topical steroids.
CSCR occurs most often in males between 20 and 50 years of age. It usually presents with acute onset of unilateral metamorphopsia, blurred vision and a relative scotoma. Patients can also experience micropsia, impaired dark adaptation, and color desaturation. Presenting visual acuity ranges from 20/15 to 20/200 with an average of 20/30. On fundus examination, there is often an elevation of the neurosensory retina in the central macula due to subretinal fluid. It is important to perform careful inspection of the fundus to exclude conditions such as optic nerve pit, age-related macular degeneration, polypoidal choroidal vasculopathy, uveitis, and intraocular tumor which can also cause subretinal fluid. OCT can be used to identify and follow the amount of subretinal fluid.1, 2 FA reveals leakage of dye from a focal choroidal/RPE defect which collects in the subretinal space. This usually occurs in an ink-blot pattern in which the spot of hyperfluorescence secondary to leakage spreads concentrically, but in 10-15% of cases, the dye can spread linearly in a smoke stack configuration, as demonstrated in the imaging from the patient described here.
Visual disturbances often resolve spontaneously in a few months, although recurrence is common and chronic CSCR can occur. Some patients experience permanent visual changes. If the subretinal fluid fails to resolve with observation alone, photodynamic therapy (PDT) may be performed to hasten subretinal fluid resorption. There is no evidence that PDT improves final visual outcomes, but since photoreceptor atrophy begins as early as 4 months after onset, there may be a theoretical a long term benefit to treatment if the fluid persists for more than 3 months. However, PDT is not without significant risk of vision loss and secondary CNV. The use of half-dose verteporfin and a 10 minute time interval between infusion and laser application may reduce these risks while still providing efficacious treatment of the abnormal choroidal vasculature.
In additional to PDT, several other treatment options for CSCR have been studied. However, studies with large sample sizes and prospective, controlled design are lacking. Studies of the efficacy of bevacizumab and ranibizumab therapies have shown significantly improved retinal thickness and visual acuity in the first 3 months following treatment, but this improvement is not sustained beyond 6 months.3-7
Medications that inhibit mineralocorticoid production or antagonize the mineralocorticoid receptor have recently been studied as potential treatments for CSCR. Zhao et al.8 demonstrated efficacy of eplerenone in a CSCR animal model, and a small pilot study of 13 human patients showed promise.9. There are a handful of ongoing clinical trials for eplerenone, which is an FDA-approved medication used in the treatment of congestive heart failure.
Caccavale et al.10 studied the efficacy of aspirin for CSCR treatment with the rationale that aspirin may correct abnormalities of the choroidal vasculature and reduce serum levels of plasminogen activator inhibitor 1 (PAI-1), elevated levels of which have been found in patients with CSCR.11, 12 Caccavale et al. conducted a prospective trial in which patients treated with six months of 100 mg aspirin demonstrated improved visual acuity compared to historic controls. Significant improvements in visual acuity were present at one month and three months, and persisted to twelve months.
In the case of the patient described in this case report, she was eagerly seeking some kind of treatment. PDT was not recommended because at one month after onset, the risk of vision loss from PDT was not justified in a disease with a 90% spontaneous resolution rate. Eplerenone was considered, but excluded because of the need for monitoring serum potassium. The patient was started on low dose aspirin since there was some evidence supporting its efficacy, and the risks of this treatment were relatively low.
Series: Case Report 2015
References:
- Kanski J, Bowling B. Clinical Ophthalmology, 7 ed. United Kingdom: Elsevier Health Sciences, 2011.
- Ryan S, Schachat A, Wilkinson C, Hinton D, Sadda S, Wiedemann P. Retina, 5 ed. United Kingdom: Elsevier Health Sciences, 2013.
- A randomized trial comparing intravitreal triamcinolone acetonide and focal/grid photocoagulation for diabetic macular edema. Ophthalmology 2008;2008 Sep;115:1447-9.
- Aydin E. The efficacy of intravitreal bevacizumab for acute central serous chorioretinopathy. J Ocul Pharmacol Ther 2013;29:10-3. doi: 10.1089/jop.2012.0072. Epub 2012 Aug 27.
- Lim JW, Ryu SJ, Shin MC. The effect of intravitreal bevacizumab in patients with acute central serous chorioretinopathy. Korean J Ophthalmol 2010;24:155-8. doi: 10.3341/kjo.2010.24.3.155. Epub 2010 Jun 5.
- Artunay O, Yuzbasioglu E, Rasier R, Sengul A, Bahcecioglu H. Intravitreal bevacizumab in treatment of idiopathic persistent central serous chorioretinopathy: a prospective, controlled clinical study. Curr Eye Res 2010;35:91-8. doi: 10.3109/02713680903428306.
- Park SU, Lee SJ, Kim M. Intravitreal anti-vascular endothelial growth factor versus observation in acute central serous chorioretinopathy: one-year results. Korean J Ophthalmol 2014;28:306-13. doi: 10.3341/kjo.2014.28.4.306. Epub 2014 Jul 22.
- Zhao M, Celerier I, Bousquet E, et al. Mineralocorticoid receptor is involved in rat and human ocular chorioretinopathy. J Clin Invest 2012;122:2672-9. doi: 10.1172/JCI61427. Epub 2012 Jun 11.
- Bousquet E, Beydoun T, Zhao M, Hassan L, Offret O, Behar-Cohen F. Mineralocorticoid receptor antagonism in the treatment of chronic central serous chorioretinopathy: a pilot study. Retina 2013;33:2096-102.
- Caccavale A, Romanazzi F, Imparato M, Negri A, Morano A, Ferentini F. Low-dose aspirin as treatment for central serous chorioretinopathy. Clin Ophthalmol 2010;4:899-903.
- Yamada R, Yamada S, Ishii A, Tane S. [Evaluation of tissue plasminogen activator and plasminogen activator inhibitor-1 in blood obtained from patients of idiopathic central serous chorioretinopathy]. Nihon Ganka Gakkai Zasshi 1993;97:955-60.
- Iijima H, Iida T, Murayama K, Imai M, Gohdo T. Plasminogen activator inhibitor 1 in central serous chorioretinopathy. Am J Ophthalmol 1999;127:477-8.
Identifier: Moran_CORE_21271
Copyright statement: Copyright 2015. Please see terms of use page for more information.
The Not So Incidental Finding: A Cavernous Hemangioma
Home / Ophthalmic Pathology / Additional Resources
 Loading...
Loading...
Title: The Not So Incidental Finding: A Cavernous Hemangioma
Authors: Christopher D. Conrady, MD, PhD, Jun Guan, MD, H. Christian Davidson, MD, and Bhupendra Patel, MD
Date: 09/28/2015
Keywords/Main Subjects: Cavernous hemangioma; Orbit; Orbital mass; Proptosis
Secondary CORE Category: Orbit, Eyelids and lacrimal System / Orbital Neoplasms and Malformations / Vascular Tumors, Malformations, and Fistulas
Diagnosis/Differential Diagnosis: cavernous hemangioma
Description: Thirty-one-year-old female with past medical history significant for a traumatic brain injury, von Willebrand disease, and migraines, that presented to an outside ophthalmologist with recurrent headaches and “sparkles within her vision.”. She was noted to have a normal exam, including dilated fundus exam, with one exception, mild disk edema of the left optic nerve. Humphrey visual fields were normal. The patient was referred for imaging due to a change in headaches and associated visual symptoms not typical of prior migraines. MRI of the orbits identified a well-circumscribed mass within the left orbit abutting the globe (Figure 1).
The patient was referred to the oculoplastics service at the Moran for further evaluation and treatment due to concern of optic nerve compression with edema on exam. She was found to 20/15 vision, pupils were equal and reactive without relative afferent defect, extraocular motility full, and color vision was normal OU. However, there was noticeable periorbital asymmetry with increased fullness of the left lateral orbit compared to right and left-sided proptosis of 1.5 mm.
Due to concern of optic nerve changes, the patient underwent an anterior orbitotomy with complete resection of the orbital mass without complications. Histopathological specimens were consistent with an orbital cavernous hemangioma with a pseudocapsule and large vascular spaces lined with endothelium (Figure 2). The patient made a quick and full recovery with a resolution of symptoms.
Images:

Figure 1: Well-circumscribed left orbital mass on MRI of the orbits. (a) Axial MRI of the orbits with contrast with large, well-circumscribed, enhancing lesion of the left orbit. (b) Time course of enhancement showing fill-in of lesion. Radiographic findings are typical for cavernous hemangioma.

Figure 2: Cavernous hemangioma on histopathology. (a) Gross surgical specimen of the cavernous hemangioma. H&E staining was then performed for further evaluation with (b) 20x, (c) 40x, and (d) 100x images. Pseudocapsule and vascular spaces lined with endothelium are noted.
Summary of Case:
- Consider cavernous hemangiomas in both men and women with painless, unilateral proptosis in the 3rd to 5th decade of life.
- Use imaging to help aide in diagnosis as carvernous hemangiomas are well-circumscribed intraconal masses of which 90% can be diagnosed pre-operatively.
- Multiple emerging ways to access orbital tumors that decrease post-operative morbidity.
- Hormones (i.e. pregnancy and menopause) may influence size of lesion but no known factors predictive of growth.
- Presenting symptoms: Eyeball protrusion (90%), visual impairment (65%), double vision (20%), local Pain (18%), headache (12%), eyelid fullness/swelling (5%).
- For more details on the case, please see associated PowerPoint Presentation.
Format: PowerPoint presentation
References:
- Di Tomasso et al., Progesterone receptor expression in orbital cavernous hemangioma. Virchows Arch. 2000.
- Fries and Char, Bilateral orbital cavernous hemangioma. Br. J. Ophtho. 1988.
- Goldberg et al., Orbital Tumors Excision without bony marginotomy under local and general anesthesia. J. Ophtho. 2014.
- Hsu and Hsu, Cavernous Hemangioma of the Orbit: 42 patients. J. Exp and Clinc Med, 2011.
- Jayaram et al., Potential correlation between menopausal status and the clinical course of orbital cavernous hemagniomas. Optho Plast Recon Surg. 2015.
- Paluzzi et al., “Round-the-Clock” Surgical Access to the Orbit. J. Neurol Surg, 2015
- Som and Curtin., Head and neck imaging. Mosby. ISBN: 0323009425
Faculty Approval by: Dr. Bhupendra Patel, MD
Identifier: Moran_CORE_20295
Christopher D. Conrady, ©2015. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Disclosure: The authors have no financial conflicts of interest.
Case Report of Susac Syndrome
Home / Pediatric Ophthalmology and Strabismus / Uveitis in the Pediatric Age Group
Title: Case Report of Susac Syndrome
Author(s): Russell Swan MD, Rachael Jacoby MD
Date: 04/02/2015
Secondary CORE Category: Neuro-ophthalmology / Causes of Decreased Vision / Vascular Disorders
Keywords / Main Subjects: Susac syndrome; Branch retinal arterial occlusion
Diagnosis: Susac syndrome
Differential: Vasculitis, Churg Strauss, SLE, Sarcoid, Behcets, Eales, Lyme, Syphilis, TB, viral encephalitis, primary CNS lymphoma, MELAS, isolated BRAO
Brief Description: A twenty-seven year old female presents with one week of blurry vision in left eye. She has also had two weeks of dizziness and confusion; she presented to an outside hospital with inability to perform activities of daily living and not oriented to date. She was then transferred for further care at our facility. MRI showed multiple subcortical lesions in multiple vascular territories most noticeably in the corpus collosum.
Our patient was treated with Cellcept and high dose steroids; at her follow up visit she was on Prednisone 40 mg per day and 1000 mg of Cellcept. She will be followed by neurology with serial MRIs and ophthalmology with repeat wide field fluorescein angiogram to monitor for recurrent vascular occlusions.
Further systemic evaluation included:
- TEE: no vegetation, right to left PFO
- Doppler of legs: no DVT
- CTA: normal without stenosis
- Cerebral angiogram: no stenosis or vasculitis
Labs: normal CBC, CMP, negative cardiolipin antibody, normal CRP, normal A1C, normal lupus anticoagulant, negative Factor V, Protein C/s, antithrombin III, normal homocysteine, ACE, ANCA, SSA/SSB, ANA, RF, ESR;
Lumbar puncture: normal, no oligoclonal bands.
Summary of the Case: Susac syndrome is an autoimmune disease first described in 1979 by Dr. John Susac. Most often occurs in females between the ages of 20 to 40 with a 3:1 female to male ratio. Susac syndrome typically causes visual field changes, hearing loss and vertigo, headaches with associated vomiting, confusion and cognitive difficulties. The pathophysiology is unknown at this time. Treatment includes steroids, immune modulating treatments such as cyclophosphamide, mycophenaloate, azathioprine and newer biologics, treatment can also include IVIG. The disease process is usually self-limited, lasting between 2 -4 years. The prognosis is varied, as some patients do quite well with limited treatment while others might have a recurrent disease process. Some patients develop long term cognitive deficits, gait disturbances as well as hearing and vision loss.
References:
• Egan, R., Gass, J. et al. Retinal arterial wall plaques in Susac Syndrome. American Journal Ophthalmology 2003, April 135 (4) 483-486
• Susac, J. Susac’s Syndrome. American Journal of Neuroradiology 2004, 25:352-352
Identifier: Moran_CORE_314
Images:
Fundus photos of right eye are normal, left eye shows retinal whitening in the distribution of the superotemporal artery

Early phase of the fluorescein angiogram in the left eye shows delayed arterial filling in the superotemporal artery

Follow up wide field FA three months later shows continued delayed arterial filling in the superotemporal artery branch

Late phase of the follow up angiogram showing vessel wall staining in the inferotemporal vessels.
Copyright statement: Copyright ©2015. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Case Report of Non-Arteritic Ischemic Optic Neuropathy (NAION)
Home / Neuro-Ophthalmology / Grand Rounds Presentations and Cases
Title: Case Report of Non-Arteritic Ischemic Optic Neuropathy (NAION)
Author (s): Eileen Hwang, Judith Warner
Photographer (s): Mel Chandler, Cyrie Fry
Date: 05/08/15
Keywords/Main Subjects: NAION, non-arteritic ischemic optic neuropathy, visual field defect, optic disc edema, optic nerve edema, optic neuropathy, afferent pupillary defect
Diagnosis/Differential Diagnosis: non-arteritic ischemic optic neuropathy
Brief Description:
Chief complaint: visual field defect
History of present illness: The patient is a 73-year-old woman who presented two days after sudden onset of vision changes. She described a gray film in the inferior area of the vision in her right eye that had not changed since onset. The patient denied having fevers, chills, weight loss, jaw claudication, shoulder stiffness and weakness. Her medical history was significant for hypertension, hyperlipidemia, and rheumatoid arthritis, and her medications included enalapril 20 mg twice a day, hydrochlorothiazide 10 mg daily in the morning, and aspirin 81 mg daily. She was in the habit of measuring her blood pressure daily and reported that it was consistently in the 130s/50s-60s. She denied feeling lightheaded when standing up. She denied snoring.
Initial examination: The patient’s visual acuity was 20/20 in each eye with pin hole and she did not have a relative afferent pupillary defect. She had mild elevation without edema of the superior optic disc in the right eye and a retinal nerve fiber layer hemorrhage nasal to the disc (Figure 1). Her cup-to-disc ratio in the right eye was 0.2. The cup-to-disc ratio in her left eye was 0.6. The patient was referred to glaucoma, and subsequently to neuro-ophthalmology.
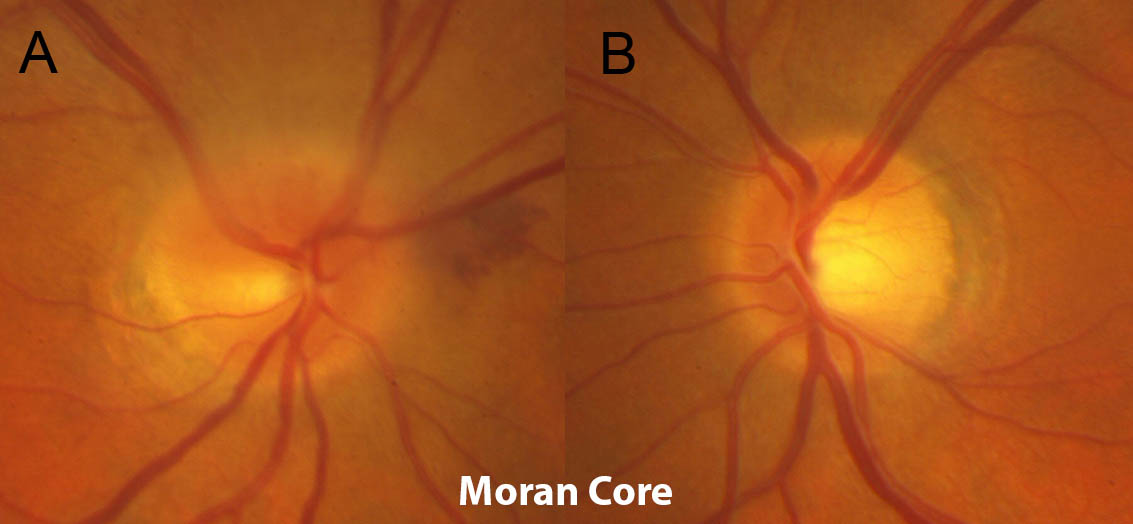
Figure 1. Optic disc photos from the patient’s initial examination. A) The superior part of the right optic nerve is mildly elevated and there is a peripapillary hemorrhage nasally. There is a cup to disc ratio of 0.2. B) The left optic nerve is without elevation or edema and has a cup to disc ratio of 0.6.
Follow up examination in the neuro-ophthalmology clinic: Two weeks later, the patient’s visual acuity with pinhole was 20/20 in the right eye and 20/20 in the left eye. She had a 0.6 log unit right relative afferent pupillary defect. Her color vision was decreased on the right with 80% red desaturation and 11/13 Ishihara plates compared to 13/13 on the left. Her critical flicker fusion frequency was 39 Hz on the right and 41 Hz on the left. On slit lamp examination, there was 350 degrees of disc edema in the right eye with obscuration of the nasal vessels. There were also exudates on the nasal disc and a retinal nerve fiber layer hemorrhage nasal to the disc (Figure 2). On Humphrey visual field testing, there was an inferior arcuate defect in the right eye. The left eye visual field showed an inferior rim artifact (Figure 3).

Figure 2. A) Infrared image and B) OCT RNFL showing 350 degrees of optic nerve edema with obscuration of the nasal vessels of the right eye on two week follow up examination.
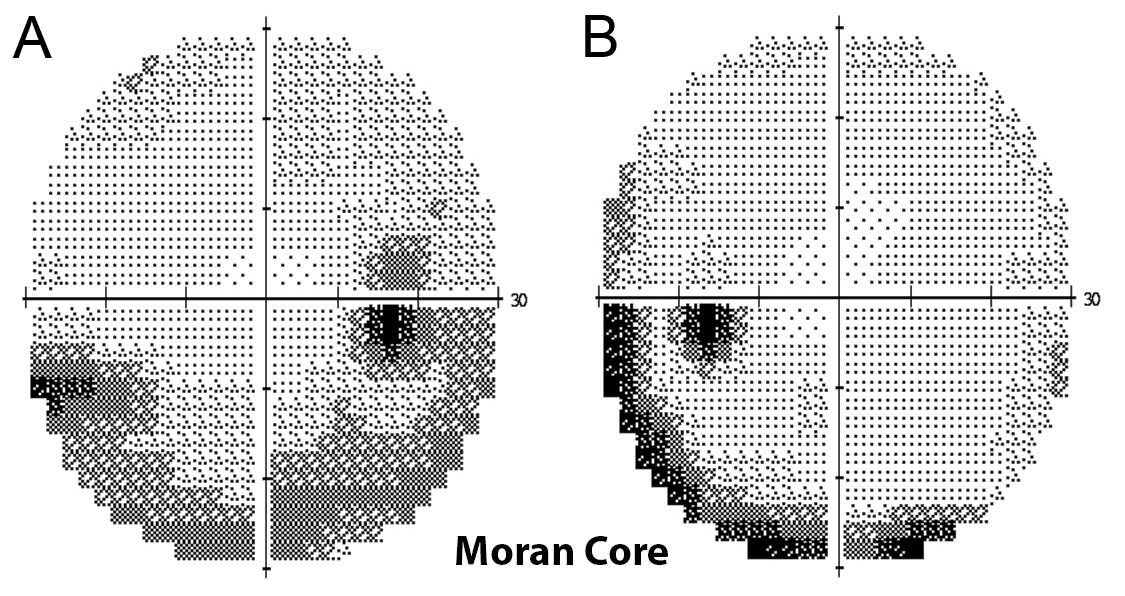
Figure 3. Humphrey visual field 30-2 demonstrating A) inferior arcuate defect of the right eye and B likely rim artifact of the left eye.
Clinical course: The patient was diagnosed presumptively with non-arteritic ischemic optic neuropathy although other possibilities that were considered were a compressive mass lesion, central retinal vein occlusion, diabetic papillitis, hematological abnormalities (thrombocytosis, anemia), vasculitis, and hypertensive papillitis. Lab tests were performed to rule out giant cell arteritis, and causes of hypercoagulability such as thrombocytosis, polycythemia, and multiple myeloma. The results of these laboratory studies (erythrocyte sedimentation rate, c-reactive protein, complete blood count, and serum protein electrophoresis) were normal.
The patient decided to start brimonidine twice daily for neuro-protection after an informed discussion about the limited evidence supporting its use. In conjunction with her primary care doctor, she decreased her enalapril to once a day in the morning. Her diastolic blood pressure remained in the 50s-60s.
Four weeks after initial symptom onset, the patient reported that the visual field defect had suddenly extended to include part of her central vision. Her visual acuity was stable, but the magnitude of her right relative afferent pupillary defect increased to 0.9 log units and her color vision worsened to 5/13 Ishihara plates. On slit lamp examination, her optic nerve edema had almost completely resolved except for mild blurring of the margin inferiorly. Humphrey visual field testing demonstrated a new superior paracentral scotoma with improvement of the inferior altitudinal defect. Due to the unusual nature of her late progression and the lack of a “disc at risk” (i.e. small cup to disc ratio) in the contralateral eye, the decision was made to order cerebral imaging to rule out a compressive or infiltrative lesion.

Figure 4. Follow up A) OCT RNFL and B) infrared image demonstrating resolution of the optic nerve edema of the right eye.

Figure 5. Humphrey visual field 30-2 testing performed after the patient noticed additional subjective field changes four weeks after initial symptom onset demonstrating A) a new superior paracentral visual field defect of the right eye and B) full field in the left eye.
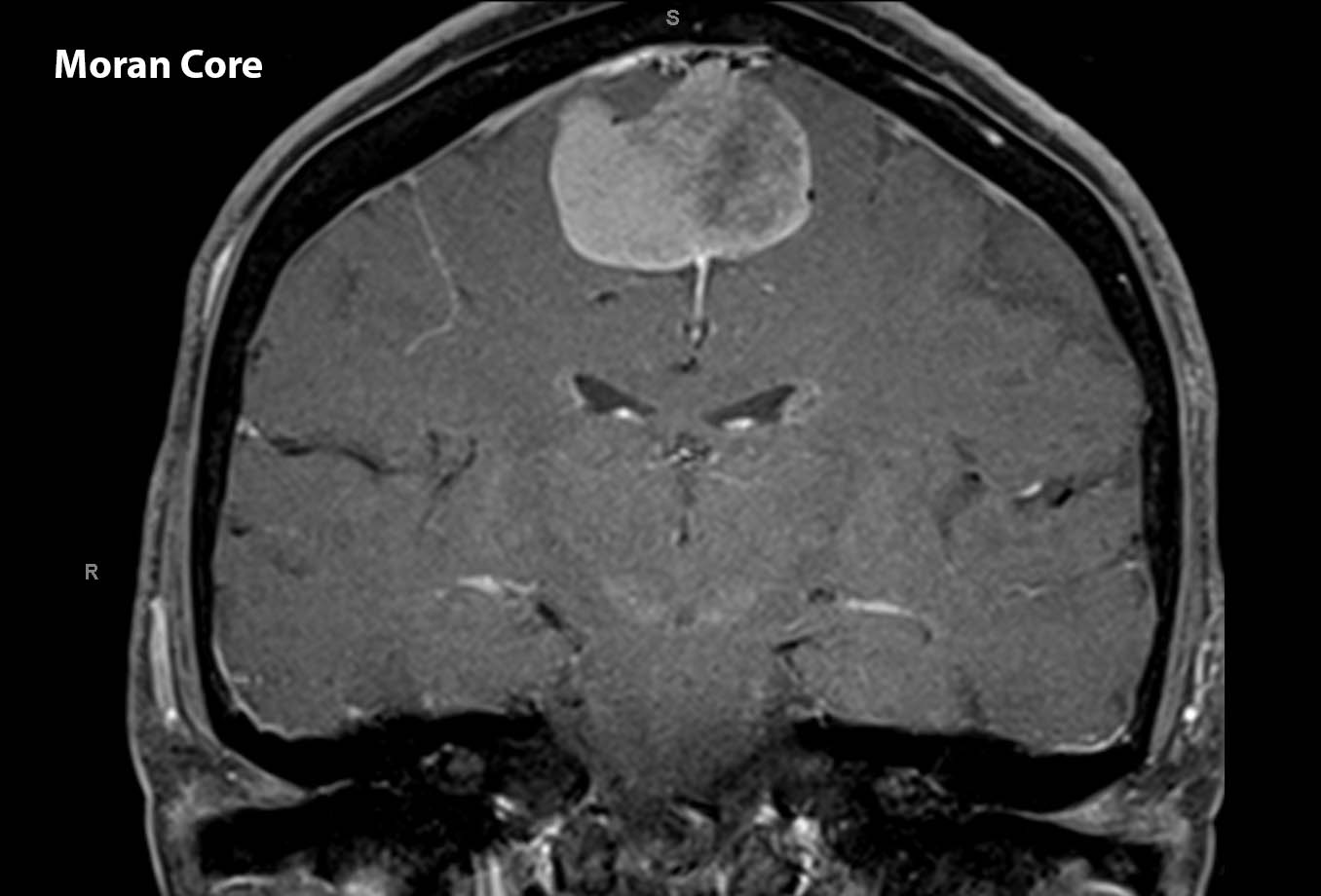
Figure 6. MRI of the brain coronal T1 post contrast demonstrating a sagittal lesion with characteristics consistent with meningioma.
The MRI demonstrated a sizeable sagittal lesion. The patient was referred to neurosurgery and had the lesion resected. It was confirmed by pathology to be a mengioma, WHO grade I. The patient was able to return to her work as a full time nurse in the cardiac catheterization lab one week after surgery without any neurological deficits.
Discussion:
Anterior ischemic optic neuropathy (AION) is characterized by rapid onset of unilateral vision loss associated with optic nerve edema. There are two types of anterior ischemic optic neuropathy – arteritic anterior ischemic optic neuropathy (AAION), and non-arteritic anterior ischemic optic neuropathy (NAION). To make a diagnosis of NAION, AAION should be sufficiently ruled out, which can be by a history negative for symptoms of temporal arteritis (headache, scalp tenderness, jaw claudication, fever, weight loss, and proximal joint pain) and young age (less than 50 years old), but also by laboratory testing or temporal artery biopsy, depending on the level of clinical suspicion. In the evaluation of a patient with acute onset of unilateral vision changes and optic nerve edema, optic neuritis must also be considered, and characteristics such as a younger patient age and pain with eye movements are suggestive of optic neuritis rather than NAION.
After acute onset, the vision loss usually remains static, although it can also progress in either a gradual or stepwise fashion for a few weeks before stabilizing. On examination, there is optic nerve edema that can be segmental and usually is not chalky white. Peripapillary hemorrhages are common. The optic nerve in the contralateral, unaffected eye usually has a small cup-to-disk ratio, and this is thought to contribute to risk of NAION because the crowding supports propagation of ischemia and swelling. Visual field defects due to NAION are most commonly altitudinal, although many other types of defects can be present as well.
Fluorescein angiography in patients with acute NAION demonstrates delayed filling of the prelaminar optic disc, supporting the role of anterior ischemia in the pathogenesis of NAION. Possible risk factors for NAION that may exacerbate optic nerve ischemia include nocturnal hypotension and sleep apnea. Recently, it has been demonstrated that erectile dysfunction medications increase the risk of NAION, although this is a very rare event (Campbell, 2015). After NAION has been diagnosed, interventions should focus on minimizing risk factors, since there are no acute interventions that have been shown to improve outcomes. There is poor evidence that brimonidine eye drops or oral low-dose aspirin can prevent future events in the contralateral eye (Kupersmith, 1997), but at our institution, brimonidine is usually started in both eyes for its neuroprotective effect. It is usually stopped in the acutely affected eye after the optic nerve edema has resolved since that disc is no longer at risk for NAION secondary to atrophy but continued in the unaffected eye. Low dose aspirin is usually recommended as well.
Visual acuity and visual field defects can improve somewhat in the months following the initial onset, but usually do not return to baseline. There is rarely ever progression beyond 2 months, and therefore any evidence of progression should be an indication to look for other causes of unilateral optic nerve edema, such as a mass lesion or intraocular inflammation.
References:
Miller NR, Newman NJ, Biousse V, Kerrison JB, Editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology. Lippincott Williams & Wilkins, Philadelphia, 2005.
Campbell UB, Walker AM, Gaffney M, Petronis KR, Creanga D, Quinn S, Klein BE, Laties AM, Lewis M, Sharlip ID, Kolitsopoulos F, Klee BJ, Mo J, Reynolds RF. Acute nonarteritic anterior ischemic optic neuropathy and exposure to phosphodiesterase type 5 inhibitors. J Sex Med. 2015 Jan;12(1):139-51.
Kupersmith MJ, Frohman L, Sanderson M, Jacobs J, Hirschfeld J, Ku C, Warren FA. Aspirin reduces the incidence of second eye NAION: a retrospective study. J Neuroophthalmol. 1997 Dec;17(4):250-3.
Identifier: Moran_CORE_275
Copyright statement: Copyright ©2015. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/