
Retinoblastoma
Home / Ophthalmic Pathology / Additional Resources
Title: Retinoblastoma
Author: Amber Jimenez
Photographer: Moran Eye Center photography
Date: 10/17/2019
Keywords/Main Subjects: tumor, tumor suppressor, leukocoria, RB1
Diagnosis: Retinoblastoma
Brief Description:
Retinoblastoma is an intraocular malignancy of the retinal cells resulting from a disruption of a tumor suppressor gene, called RB1. The disease often presents in children within the first two years of life. The most common symptom of retinoblastoma is leukocoria, or white pupil. One, or both eyes may be affected, and extent of the disease may vary, depending largely on the type of mutation being germline or somatic. Left untreated, retinoblastoma may have debilitating effects on vision and can be fatal. Prompt referral for management of the condition is recommended.
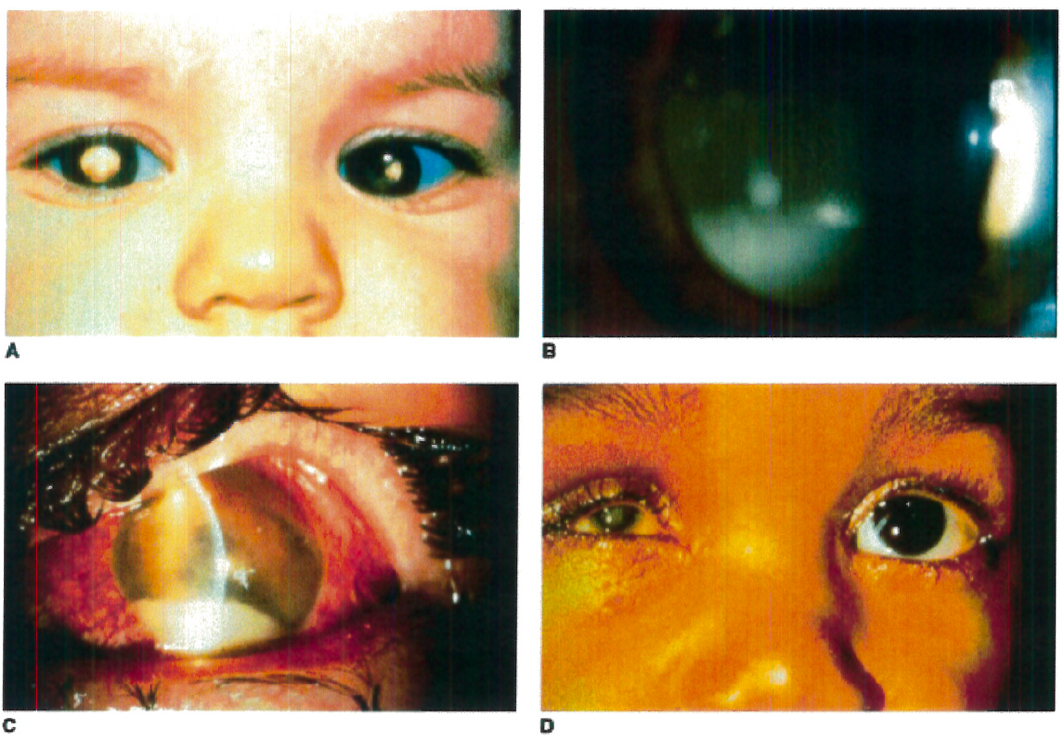
Figure 1. A: Heritable RB patient presenting with bilateral leukocoria, the most commonly reported presenting sign of this condition. B: RB cells have a propensity to detach from the primary tumor mass and spread throughout the eye, as illustrated by a patient with endophytic RB who developed marked tumor cells in the vitreous, resembling vitritis. C: Occasionally tumor necrosis may produce an inflammatory response associated with a red eye and conjunctival injection as seen in this patient. The patient also has a tumor hypopyon. D: Massive tumor necrosis in the phthisical eye of a patient with RB has resulted in marked inflammation involving the orbit, simulating an orbital cellulitis with lid swelling and pain on eye movement.
Taken from Figure 26.4 A-D, Hartnett, M.E. (2014) Pediatric Retina. Philadelphia, PA. Lippincott Williams & Wilkins, a Wolters Kluwer business.

Taken from Figure 26.5 B, Hartnett, M.E. (2014) Pediatric Retina. Philadelphia, PA. Lippincott Williams & Wilkins, a Wolters Kluwer business.
Age & Gender: The incidence of retinoblastoma is similar in males and females. Most cases are diagnosed at birth or early infancy, with an average age of diagnosis of bilateral disease at 12 months and 24 months for patients with unilateral disease.
Genotype: Both heritable (germline mutation) and nonheritable (somatic mutation) retinoblastoma arise from mutations in the retinoblastoma gene, RB1, on both alleles of chromosome 13q14.
Symptoms: The most common clinical presentation of retinoblastoma includes symptoms of leukocoria, white pupil, or strabismus in a child under 2 years of age. Other symptoms include nystagmus or a red inflamed eye.
Environmental Factors or Other Considerations: Germline mutations of RB1 have been found to occur on paternally derived chromosomes, suggesting that increased paternal age may have impact on the occurrence of retinoblastoma. No other environmental factors have been identified.
Treatment: Treatment options vary depending on the size and location of the tumor, extent of visual disturbance, optic nerve involvement, and presence of metastatic disease. Surgery, brachytherapy or radiation, chemotherapy, and laser therapy are common treatments.
Pathophysiology: The RB1 gene encodes the Rb protein which functions as a tumor suppressor by restricting progression of the cell cycle in dividing cells. Loss of function in both copies of RB1 causes cell cycle dysregulation, allowing for potentially cancerous cells to continue dividing. The Knudson “two-hit” model differentiates the heritable and nonheritable forms. Patients with the heritable form of retinoblastoma inherit a germline mutation of RB1 in all cells in the body, thus creating the “first hit.” A “second hit” occurs when the second allele of RB1 is mutated or silenced through epigenetic changes, affecting retinal cells. Patients with heritable retinoblastoma are at increased risk for bilateral and multifocal disease. Nonheritable retinoblastoma occurs with somatic mutations of both copies of the RB1 gene (both “first” and “second” hit) in a retinal cell. These patients present with unilateral and unifocal disease, and which usually occurs later in life.
Differential Diagnosis: The most common differential diagnoses include conditions associated with the most common presenting symptom of retinoblastoma, infantile leukocoria. Such conditions include congenital malformations such as persistent fetal vasculature, vascular changes such as Coat’s disease, and inflammatory diseases such as ocular tococariasis. A patient’s age, combined with a consistent clinical presentation, morphologic features, and family history, are key to differentiating these conditions from retinoblastoma.
References: Originally published in: Hartnett, Mary Elizabeth. Pediatric retina: medical and surgical approaches. 2nd Ed. Philadelphia: Lippincott Williams & Wilkins, 2005.
Abramson, D. H., Frank, C. M., Susman, M., Whalen, M. P., Dunkel, I. J., & Boyd, N. W. (1998). Presenting signs of retinoblastoma. The Journal of Pediatrics,132(3), 505-508. doi:10.1016/s0022-3476(98)70028-9
Abramson, D. H., Beaverson, K., Sangani, P., Vora, R. A., Lee, T. C., Hochberg, H. M., Kirszrot, J., Ranjithan, M. (2003). Screening for Retinoblastoma: Presenting Signs as Prognosticators of Patient and Ocular Survival. Pediatrics,112(6), 1248-1255. doi:10.1542/peds.112.6.1248
Balmer A, Munier F. Differential diagnosis of leukocoria and strabismus, first presenting signs of retinoblastoma. Clinical ophthalmology (Auckland, NZ). 2007;1(4):431-439.
Faculty Approval by: M. E. Hartnett, MD
Copyright ©2019. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/