

Limbal Stem Cell Deficiency Image Report
Home / External Disease and Cornea / Clinical Approach to Depositions and Degenerations of the Conjunctiva, Cornea, and Sclera
Title: Limbal Stem Cell Deficiency
Author: Alex Wright, BS
Date: 07/25/2017
Images:
OS: 

OD: 
Keywords/Main Subjects: Limbal Stem Cell Deficiency
Diagnosis: Limbal Stem Cell Deficiency
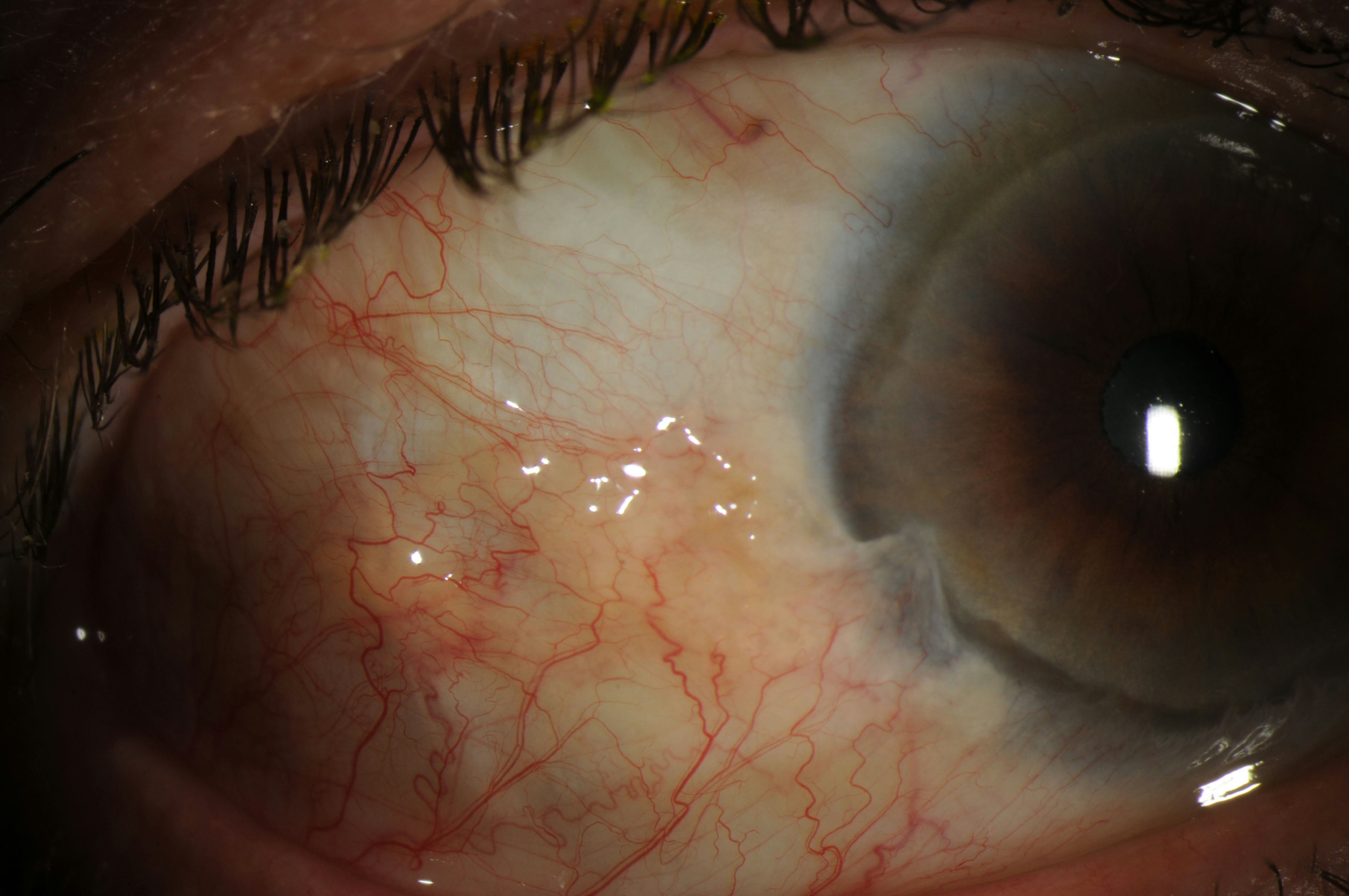
Description of Image: Limbal Stem Cell Deficiency (LSCD) occurs when the regenerative capacity of the corneal stem cells is impaired or damaged. The Palisades of Vogt, papilla-like structures of the limbus, are densely packed with basal cells, 10-15% of which are thought to be corneal stem cells. When these cells are damaged, the regenerative capacity of the cornea is gone. Thoft and Friend in 1983 originally hypothesized cell division in limbus plus further division of transit amplifying cells during their migration superficially and vertically equaled the amount of mature corneal stem cells lost and sluffed off in the tear film. As the cells are lost, the corneal surface begins to breakdown, which leads to further inflammation, conjunctival ingrowth, and subsequent neovascularization. Clinical symptoms include: photophobia, pain, conjunctivalization, corneal neovascularization, and recurrent ulceration. A diagnosis is established with clinical signs and symptoms and can be confirmed in some cases with impression cytology. Impression cytology looks for the presence of goblet cells on the cornea and helps identify conjunctivalization of the cornea, which is regarded as the most reliable diagnostic sign.
Pterygia can cause LSCD, but also must be differentiated from LSCD. Pterygia are often secondary to sun exposure and occur most commonly nasally. Pterygia are also more likely to be identified in younger patients. LSCD with pterygia are more likely bilateral and located in areas not exposed to the sun as often (notably inferiorly and superiorly). LSCD also occurs more commonly over the age of 50.
Etiologies of LSCD include: idiopathic, chemical/thermal burns, iatrogenic, autoimmune, and congenital/hereditary. Management initially if mild is aimed at controlling inflammation and the progression of the disease with topical steroids and topical cyclosporine drops. In severe cases surgery is often necessary to restore normal corneal epithelium. Many surgical procedures have been identified and studied, including: Conjunctival Limbal Autograft, Simple Limbal Epithelial Transplantation, Cultured Limbal Epithelial Transplantation, Living-Related Conjunctival Limbal Allograft, and Keratolimbal Allograft. When possible an autograft is preferred since allografts require systemic immunosuppression.
These images were taken from a 74-year-old Caucasian male with LSCD bilaterally. These images show a pterygium over the nasal cornea bilaterally and over the temporal cornea on the left eye. There is also signs of conjunctivalization and neovascularization inferiorly bilaterally.
References:
- Dua H S, Saini J S, Azuara-Blanco A, Gupta P. Limbal stem cell deficiency : Concept, aetiology, clinical presentation, diagnosis and management. Indian J Ophthalmol 2000;48:83-92
- Osei-Bempong C, Figuieredo FC, Lako M. The limbal epithelium of the eye – A review of limbal stem cell biology, disease and treatment. Bioessays 2012;35:211-219.
- Hatch KM, Dana R. The Structure and Function of the Limbal Stem Cell and the Disease States Associated With Limbal Stem Cell Deficiency. Int Ophthalmology Clin 2009;49:43-52
- Thoft RA, Friend J. 1983. The X, Y, Z hypothesis of corneal epithelial maintenance. Invest Ophthalmol Vis Sci 24: 1442–3.
- Kim KW, Mian SI. Diagnosis of Corneal Limbal Stem Cell Deficiency. CO-Ophthalmology 2017;28(4):355-362.
- Vadrevu VL, Fullard RJ. Enhancements to the conjunctival impression cytology technique and examples of applications in a clinico-biochemical study of dry eye. CLAO J 1994; 20:59–63.
- Holland EJ. Management of Limbal Stem Cell Deficiency: A Historical Perspective, Past, Present, and Future. Cornea 2015;34:S9-15.
Faculty Approval by: Dr. Brian Zaugg
Identifier: Moran_CORE_24246
Copyright statement: Copyright ©2017. Please see the Moran CORE terms of use for more information.
Disclosure (Financial or other): None
Fundus photo and OCT of Best’s vitelliform macular dystrophy (BVMD)
Home / Pediatric Ophthalmology and Strabismus / Disorders of the Retina and Vitreous
Title: Fundus photo and OCT of Best’s vitelliform macular dystrophy (BVMD)
Author (s): Gavin Gorrell, 4th Year Medical Student, University of New Mexico School of Medicine
Date: 06/24/2017
Image:

Keywords/Main Subjects: Best’s Disease; Best’s vitelliform macular dystrophy (BVMD); bestrophinopathies
Secondary CORE Category: Home / Retina and Vitreous / Hereditary Retinal and Choroidal Dystrophies
Diagnosis: Best’s vitelliform macular dystrophy (BVMD)
Description of Image:
- Egg-yolk appearance of lipofuscin accumulation at the central macula
- Subretinal fluid and lipofuscin accumulation causing RPE detachment
Pathogenesis (&Presentation): Best vitelliform macular dystrophy (BVMD) is the second most common hereditary macular dystrophy and is autosomal dominant with variable penetrance and expression. BVMD is the most common of the “bestrophinopathies”, a group of diseases which all contain various mutations to the BEST1 gene. BEST1 (previously termed VMD2), codes for bestrophin 1, a transmembrane protein in the RPE believed to be involved in anion transport and calcium signaling. Through unclear mechanisms, dysfunction of bestrophin 1 in BVMD leads to accumulation of lipofuscin (a retinal breakdown pigment) between Bruch’s membrane and RPE. This debris leads to the characteristic subretinal egg-yolk lesion.
Presentation: In early disease, usually beginning between ages 3 and 15, fundoscopy shows significant vitelliform (vitellus is latin for egg-yolk) lesions to the central macula, however patients may be asymptomatic or have only minimal loss in visual acuity. Over time the lipofuscin in the initial lesions disperses and is resorbed, leading to a “scrambled egg” appearance. A slow progression to RPE atrophy is expected with vision loss typically stabilizing around 20/30-20/200. Rapid decline in VA should cause concern for choroidal neovascularization, a feared complication that occurs in about 20% of patients (eyewiki).
Diagnosis & Differential:
Diagnosis is primarily made with family history and clinical appearance. Electro-oculography (EOG), which demonstrates abnormally low light/dark ratio in Best’s disease (<1.5), is often obtained to confirm the diagnosis. Genetic testing for BEST1 mutation is also available. Providers should consider OCT and/or fluorescein angiography to evaluate for choroidal neovascularization.
DDx: Adult-onset vitelliform macular dystrophy, toxoplasmosis retinochoroiditis, AMD, Bull’s eye maculopathy
Management: There is no current treatment available for Best’s disease, however patients should be monitored for choroidal neovascularization.
References:
Freund, K. Bailey, David Sarraf, William F. Mieler, and Lawrence A. Yannuzzi. “Hereditary Chorioretinal Dystrophies.” In The Retinal Atlas, 2nd ed., 13–231. Philadelphia, PA, 2017. https://www-clinicalkey-com.libproxy.unm.edu/#!/content/book/3-s2.0-B9780323287920000028?scrollTo=%23hl0000946.
Johnson, Adiv A., Karina E. Guziewicz, C. Justin Lee, Ravi C. Kalathur, Jose S. Pulido, Lihua Y. Marmorstein, and Alan D. Marmorstein. “Bestrophin 1 and Retinal Disease.” Progress in Retinal and Eye Research 58 (May 2017): 45–69. doi:10.1016/j.preteyeres.2017.01.006.
MacDonald, Ian M., and Thomas Lee. “Best Vitelliform Macular Dystrophy.” In GeneReviews(®), edited by Roberta A. Pagon, Margaret P. Adam, Holly H. Ardinger, Stephanie E. Wallace, Anne Amemiya, Lora JH Bean, Thomas D. Bird, et al. Seattle (WA): University of Washington, Seattle, 1993. http://www.ncbi.nlm.nih.gov/books/NBK1167/
“The Electroretinogram and Electro-Oculogram: Clinical Applications by Donnell J. Creel – Webvision.” Accessed June 24, 2017. http://webvision.med.utah.edu/book/electrophysiology/the-electroretinogram-clinical-applications/.
“Best Disease – EyeWiki.” Accessed June 24, 2017. http://eyewiki.aao.org/Best_Disease
Faculty Approval by: Griffin Jardine, MD
Disclosure (Financial or other): None
Copyright statement: Copyright 2016. Please see terms of use page for more information.
Multimodal imaging of choroidal metastases from adenoid cystic carcinoma of the submandibular gland.
Home / Ophthalmic Pathology / Ocular Involvement in Systemic Malignancies
Title: Multimodal imaging of choroidal metastases from adenoid cystic carcinoma of the submandibular gland.
Author: Brian M. Besch
Date: 06/26/2017
Images:

Keywords/Main Subjects: adenoid cystic carcinoma, choroidal metastasis, ophthalmic pathology, intraocular tumors
Diagnosis: adenoid cystic carcinoma of left submandibular gland with bilateral choroidal metastases
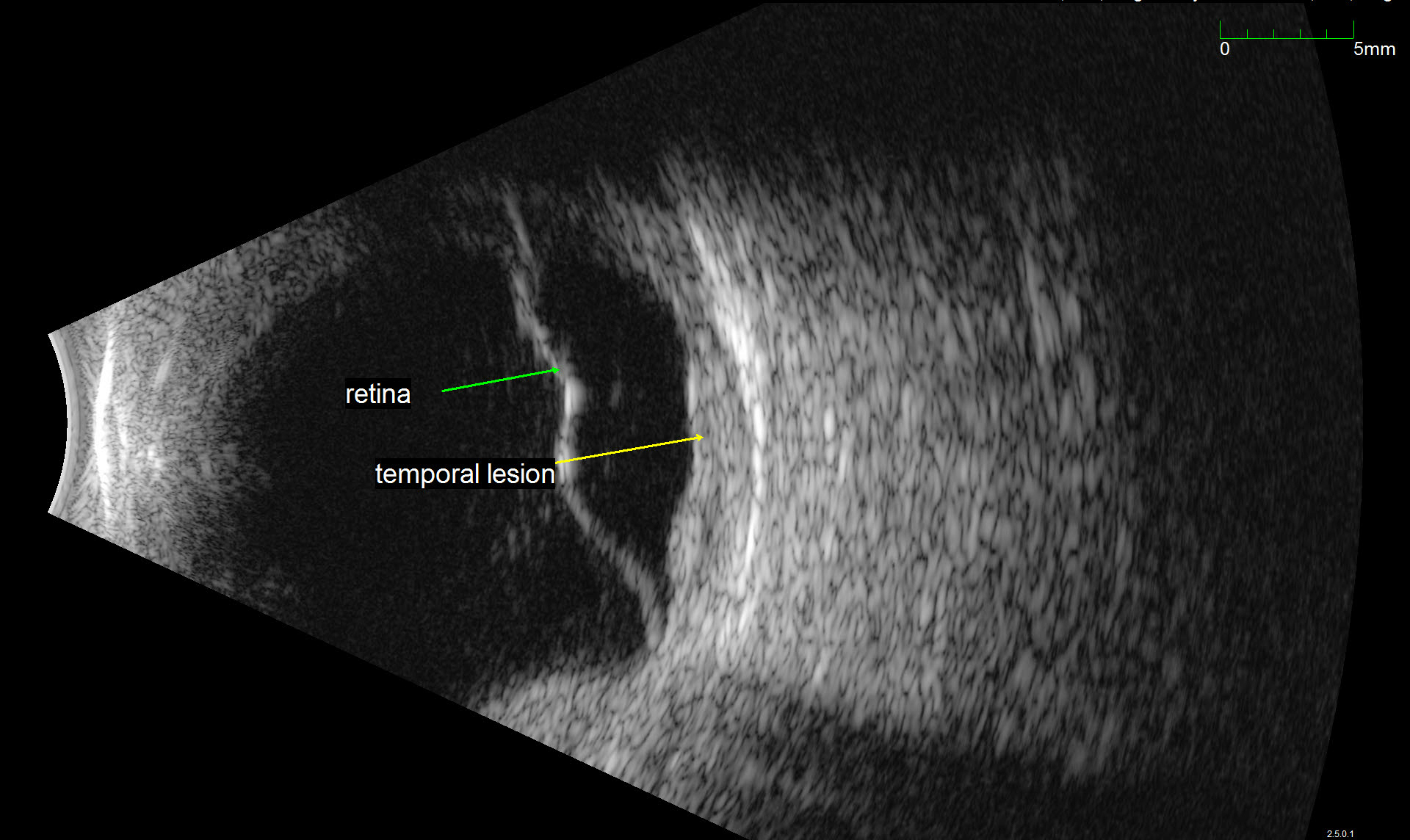
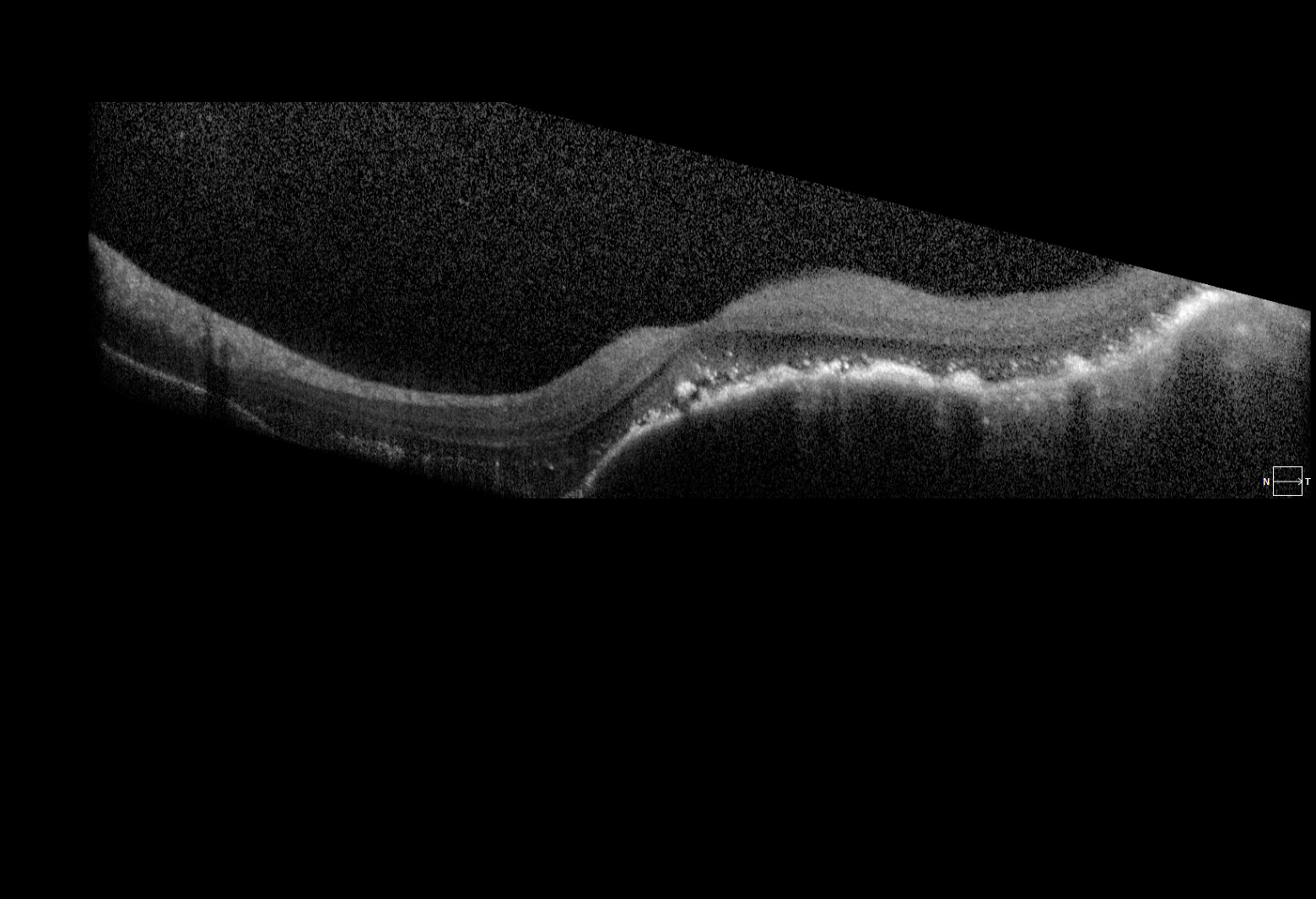
Description of Image: Adenoid cystic carcinoma (ACC) is a rare malignancy originating from multiple sites, but typically arises in the head and neck, and most commonly the salivary glands. Histologically, the lesion is composed of epithelial and myoepithelial cells in three configurations – tubular, cribiform, or solid; the latter is the most aggressive type. The malignancy generally arises between ages 40-60 and has an indolent, but nonetheless persistent course generally refractory to treatment. Initial management is wide surgical resection, typically followed by adjuvant external beam radiation or chemotherapy with cisplatin or melphalan. Distant metastases are common later in the disease course; one report indicates a mean of 48 months following initial management. Metastasis to the lung is most common. Choroidal metastases are rare; at the time of writing, the associated images represent the seventh reported case in the literature. While the limited reports of ACC in general suggest a slight female predominance, of the 6 cases involving choroidal metastases previously described, 5 were in women. All 5 primary tumors arose from submandibular glands, while the single male case derived from the hard palate.
The images represent bilateral choroidal ACC metastases from a 57 year old female. The fundus montage illustrates two metastatic lesions in the left eye; one involves the macula, and is associated with inferior serous retinal detachment. A labeled ultrasound frame indicates the larger temporal lesion and retinal detachment. The OCT scan illustrates a large choroidal mass, marked retinal architecture distortion, and sub-retinal serous edema. The patient initially presented with a lump in the submandibular region and underwent FNA with inconclusive cytology. On re-evaluation and head/neck CT imaging, she was noted to have a submandibular mass and an enlarged lymph node; she subsequently underwent left submandibular gland excision and neck dissection followed by adjuvant chemoradiation. Post-surgical CT of the chest, abdomen, and pelvis revealed multiple small pulmonary lesions bilaterally concerning for metastases. Manifestation of the choroidal lesions was noted on fundus exam approximately two years following the initial diagnosis.
References:
Cai, Qian et al. “Adenoid Cystic Carcinoma of Submandibular Salivary Gland With Late Metastases to Lung and Choroid: A Case Report and Literature Review.” Journal of Oral and Maxillofacial Surgery 72.9 (2014): 1744–1755.
Shie, Jerry A. LDS et al. “Bilateral Choroidal Metastasis From Adenoid Cystic Carcinoma of the Submandibular Gland.” Retina 20.4 (2000): 406–407.
Faculty Approval by: Griffin Jardine, MD; Akbar Shakoor, MD
Identifier: Moran_CORE_24186
Copyright statement: Copyright 2017. Please see terms of use page for more information.
Choroideremia
Home / Retina and Vitreous / Hereditary Retinal and Choroidal Dystrophies
Title: Choroideremia
Author (s): Liang Cheng, 4th Year Medical Student, University of Iowa
Photographer: Unknown
Date: 06/20/2017
Images:

Keywords/Main Subjects: Choroideremia
Diagnosis: Choroideremia
Description of Image:
Choroideremia is a rare X-linked retinal degeneration that typically affects males. It is characterized by progressive degeneration of the retinal pigment epithelium, photoreceptors, and choriocapillaris. In fact, in late stages, the underlying sclera is exposed due to complete chorioretinal dystrophy, hence the name choroid-“eremia” (Greek for bare). The disease is caused by a mutation of the CHM gene, which leads to defects in intracellular vesicular trafficking and subsequent retinal pigment epithelium (RPE), photoreceptor, and choroidal degeneration.
Patients typically present with night blindness and peripheral vision loss in teenage years. Central vision is maintained until the 5th or 6th decade of life but then can rapidly deteriorate. Carriers are mostly unaffected, but there are case reports of symptomatic female patients with less severe features. On dilated fundus exam, the earliest sign is diffuse pigment clumping, followed by atrophy with visible sclera and choroidal vessels. Atrophy progresses centripetally and the fovea is the last to become affected. Choroideremia is usually suspected based on the symptoms and findings, but a full clinical work-up includes a detailed family history, visual field testing, electroretinography (ERG), and genetic testing. Visual field testing shows patchy peripheral vision loss that corresponds to the location of the chorioretinal dystrophy, and ERG shows a reduced scotopic component early on in the disease process. Additional imaging can also be supportive of the diagnosis, such as fundus autofluorescence, infrared (IR) and optical coherence tomography. There are currently no treatments available, but gene therapy trials are underway. The differential to be considered includes retinitis pigmentosa, gyrate atrophy, and ocular albinism.
These color and IR fundus images are of a 35 year old gentleman with an established family history of choroideremia who was diagnosed with the disease in his late 20’s. The color fundus photo of his left eye shows scattered pigment clumping and diffuse atrophy. One can appreciate the bare sclera around the scalloped edge of the remaining healthy retina, as well as the visible large choroidal vessels. The IR fundus photo confirms the retinal atrophy in the periphery with the scalloped border. These findings support patient’s complaints of poor peripheral vision and trouble with night-time driving.
References:
Sorsby A, Franceschetti A, Joseph R, Davey JB. Choroideremia; clinical and genetic aspects. The British journal of ophthalmology. 1952;36(10):547-581.
Huckfeldt RM, Bennett J. Promising first steps in gene therapy for choroideremia. Human gene therapy. 2014;25(2):96-97.
Morgan JI, Han G, Klinman E, et al. High-resolution adaptive optics retinal imaging of cellular structure in choroideremia. Investigative ophthalmology & visual science. 2014;55(10):6381-6397.
Bonilha VL, Trzupek KM, Li Y, et al. Choroideremia: analysis of the retina from a female symptomatic carrier. Ophthalmic genetics. 2008;29(3):99-110.
Faculty Approval by: Dr. Bernstein, Griffin Jardine MD
Identifier: Moran_CORE_24177
Copyright statement: Copyright 2017. Please see terms of use page for more information.
Fundus Photography and Fluorescein Angiography of Susac’s Syndrome
Home / Retina and Vitreous / Other Retinal Vascular Disease
Title: Fundus Photography and Fluorescein Angiography of Susac’s Syndrome
Author: Katherine Hu, BA
Photographer: Glen Jenkins, Cyrie Frye, Danielle Princiotta
Date: 01/12/2015, 03/11/2015, 04/04/2017
Images:




Figure 1. Color fundus photography showing retinal whitening along the superior arcade with corresponding fluorescein angiography showing left superior temporal branch retinal artery occlusions. 1/12/2015.




Figure 2. Widefield fluorescein angiography OU with multiple bilateral branch retinal artery occlusions. Hyperfluorescence and vessel leakage seen in OS. 3/11/2015.


Figure 3. Widefield fluorescein angiography OU with multiple new branch retinal artery occlusions. 4/4/2017.
Keywords/Main Subjects: Susac’s syndrome, branch retinal artery occlusion (BRAO), microangiopathy
Diagnosis: Susac’s syndrome
Description of Image:
Susac’s syndrome is a rare, autoimmune microangiopathy involving the brain, retina, and inner ear. The condition can manifest clinically as a triad of encephalopathy, branch retinal artery occlusions (BRAO), and hearing loss, though not all three features may be present during the onset of the disease. Symptoms often include vision loss, headache, confusion, paranoia, vertigo, tinnitus, and deafness. The syndrome tends to occur in females between 20-40 years of age; it has not been associated with an inheritance pattern or family history. While the etiology and pathogenesis of Susac’s syndrome are unclear, the disease is thought to be an autoimmune disorder targeted at endothelial cells in the microvasculature of the brain, retina, and cochlea.
In the brain, MRI findings show distinctive white matter lesions that primarily involve the corpus callosum; abnormalities of the leptomeninges and gray matter may also be present. Audiogram can reveal sensorineural hearing loss. Fluorescein angiography is essential for identifying BRAO, pathognomonic hyperfluorescence of retinal arteriole walls, and small, yellow punctate arteriole wall plaques. Susac’s syndrome is often confused for demyelinating diseases such as multiple sclerosis and acute disseminated encephalomyelitis (ADEM). Other mimics include chronic encephalitis, meningitis, Lyme disease, thromboembolic disease, systemic lupus erythematosus, and CNS vasculitis. The disease is generally self-limiting, however, early and aggressive treatment with immunosuppressive therapy is recommended to avoid long-term sequelae from acute microvascular damage.
The color fundus photographs and fluorescein angiograms above are from patient KO, who is a 29 year-old Caucasian female. She first presented with acute headache, vertigo, worsening confusion, personality/behavioral changes, and partial vision loss in the left eye. Brain MRI showed extensive lesions in the white matter, deep gray, and cerebellar areas with corpus callosal preference, as well as scattered leptomeningeal enhancement. Neurological work up was non-focal and non-contributory. Upon examination, she was found to have a left superior temporal branch retinal artery occlusion visible on both fundus photography and fluorescein angiography (Figure 1). Fundus photography shows a central area of whitening due to ischemia as a result of microvascular occlusion. On subsequent widefield angiography, multiple bilateral BRAO with vascular leakage were seen (Figure 2). The images obtained display characteristic BRAO located distal of the arteriolar bifurcations with associated arteriolar wall plaques that do not resemble emboli or cholesterol plaques. Hyperfluorescence and leakage of the retinal arteriole wall seen are due to active endothelial injury. The patient was treated with high-dose Prednisone, Cellcept, and IVIG with relative stabilization of her disease. However, her clinical course was complicated by multiple microvascular ischemic strokes and new left-sided hearing loss in April 2017 when IVIG was discontinued. Follow-up examination revealed extensive retinal edema inferiorly in the left eye, contiguous with areas of retinal non-perfusion as a result of new BRAO suggesting progression of the disease (Figure 3).
Format: JPG file
References:
Egan RA, Hills WL, Susac JO. “Gass plaques and fluorescein leakage in Susac Syndrome.” Journal of the Neurological Sciences. 2010 Dec;299(1-2):97-100.
Rennebohm RM, Egan RA, Susac JO. “Treatment of Susac’s Syndrome.” Current Treatment Options in Neurology. 2008 Jan;10(1):67-74.
Susac JO. “Susac’s Syndrome.” American Journal of Neuroradiology. 2004 Mar; 25(3):351-352.
“Susac Syndrome.” Genetic and Rare Diseases Information Center. U.S. Department of Health and Human Services. Web. Accessed 24 June 2017. <https://rarediseases.info.nih.gov/diseases/7713/susac-syndrome>
Faculty Approval by: Akbar Shakoor, MD
Identifier: Moran_CORE_24152
Copyright statement: Copyright Conrady, ©2017. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
MEWDS (Multiple Evanescent White Dot Syndrome)
Home / Retina and Vitreous / Focal and Diffuse Choroidal and Retinal Inflammation
Title: MEWDS (Multiple Evanescent White Dot Syndrome)
Author: J. Erik Kulenkamp
Photographer: James Gilman
Date: 07/10/17
Figure 1: Montage Color Fundus Photo, Right Eye
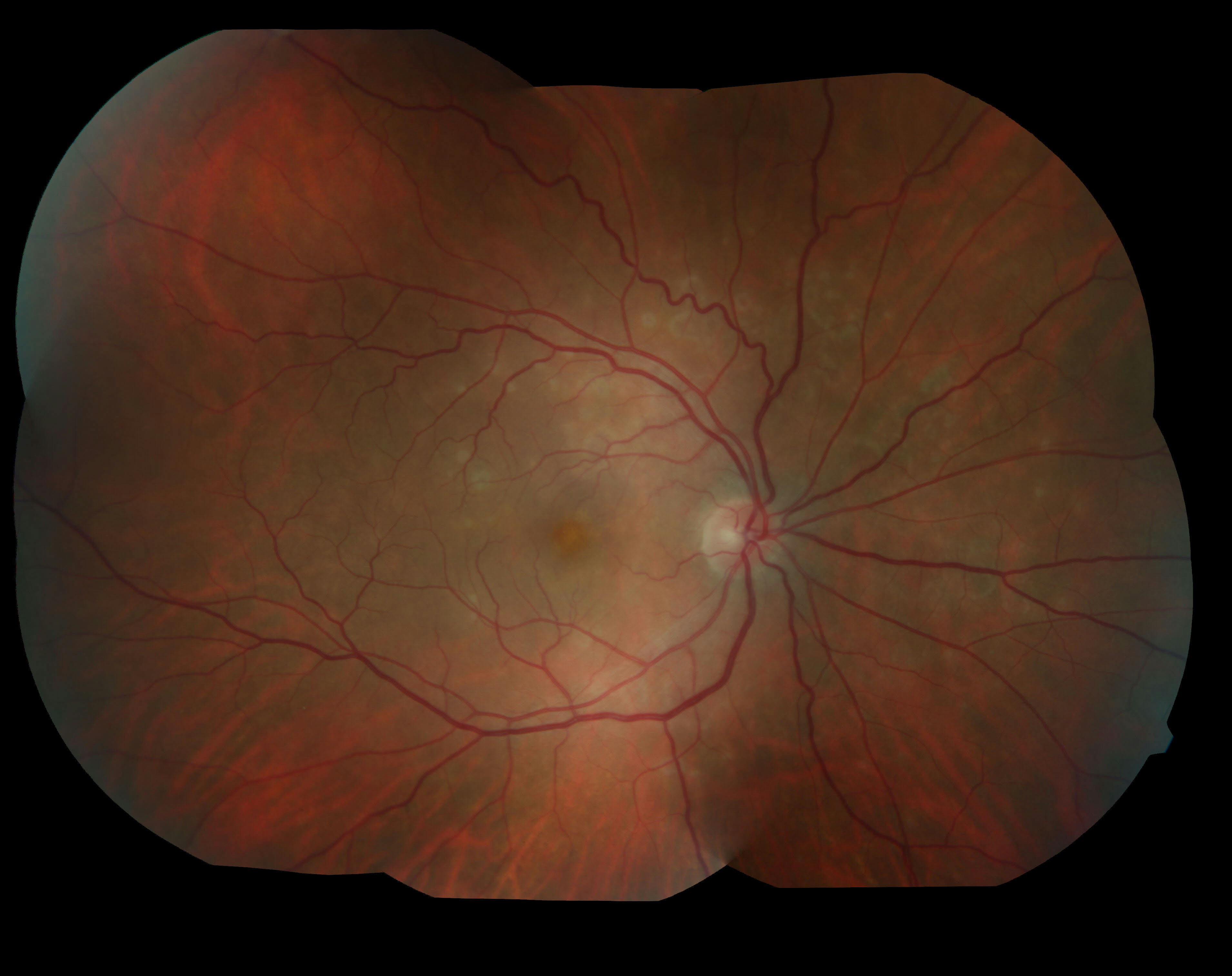
Figure 2: Fundus Autofluorescence, Right Eye
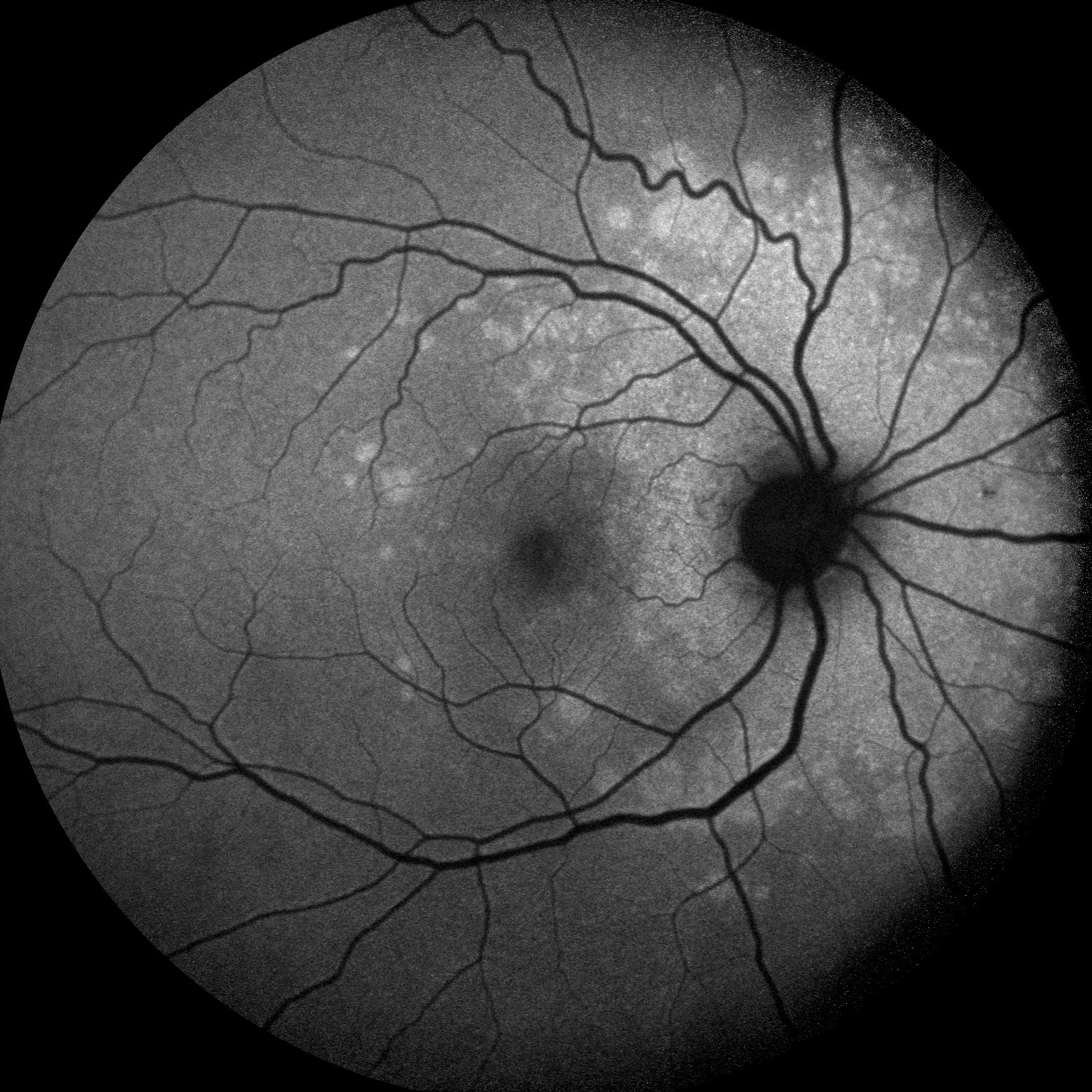
Figure 3: Fluorescein Angiogram, Right Eye

Keywords/Main Subjects: Disorders of the Retina and Vitreous – Noninfectious Retinal Inflammation
Diagnosis: MEWDS (Multiple Evanescent White Dot Syndrome)
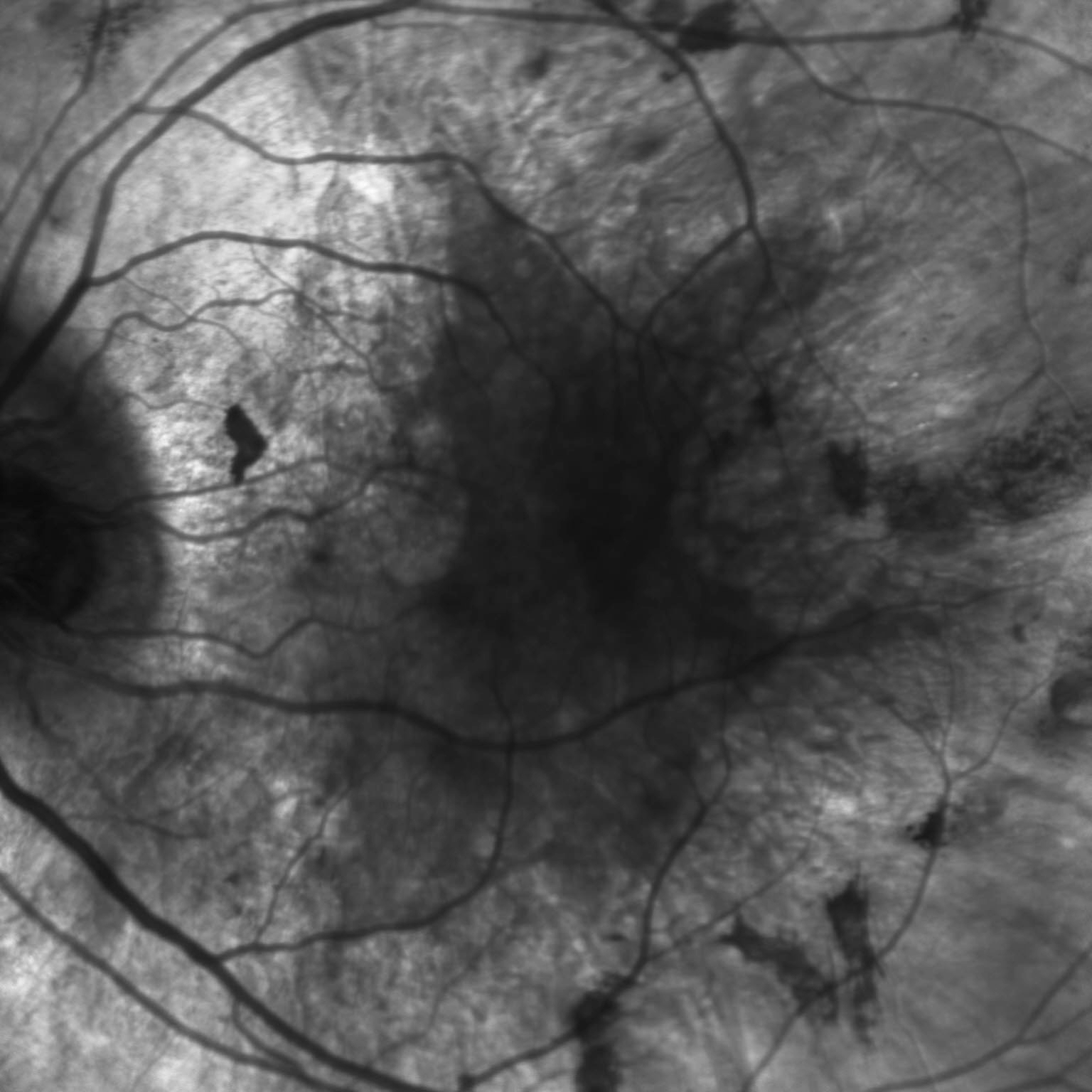
Description of Image: Multiple Evanescent White Dot Syndrome, or MEWDS, is a rare condition characterized by noninfectious inflammation at the level of the retinal pigment epithelium. It is part of a group of white dot syndromes that also includes acute posterior multifocal placoid pigment epithelipathy, birdshot retinopathy, multifocal choroiditis, multifocal choroiditis and panuveitis, punctate inner choroidopathy, and serpiginous choroidopathy1. MEWDS is much more common among females, especially those who are myopic, and typically appears in middle age. Patients usually present with decreased vision in one eye involving an enlarged blind spot, but they also occasionally experience photopsias. One-third of MEWDS cases are proceeded by a viral illness several weeks before becoming symptomatic2. When MEWDS is suspected, it is important to rule out neoplastic and infectious causes, which can sometimes share similar presentations. The characteristic finding on dilated fundoscopic examination (DFE) is small, intraretinal white dots in a wreath-like distribution around the macula3. The majority of patients improve within 2-6 weeks without steroid treatment, although the occasional individual will have a persistently enlarged scotoma2.
A 48 year old myopic female presented with one week of an enlarged blind spot. She recalls that parts of words were missing from her vision in her right eye and that the area of missing vision was surrounded by blurriness. She was born in Argentina, but denied recent travel, fevers, weight loss, shortness of breath, recent illness, pets, STI’s, or IV drug use. Visual acuity in the right eye was 20/80 -1, down from 20/50 -1 six months prior, and 20/20 in the left eye. On DFE, numerous white dots were observed in a wreath-like distribution around the macula, ranging 200-300 microns in size. These dots can be seen in Figure 1, which demonstrates the classic appearance of MEWDS in a montage color fundus photo of the right eye. The optic nerve and disc appear normal, as do the vessels, but there is foveal granularity in the macula. The white dots are densest emanating from the optic nerve. Figure 2 is a fundus autofluorescence (FAF) photo of the same eye, illustrating a multitude of hyperfluorescent dots surrounding the optic nerve and extending to encompass the macula. The extent of retinal involvement, especially nasally, is much more apparent in the FAF. Figure 3 demonstrates a fluorescein angiogram with hyperfluorescence in a similar pattern to the FAF, which remains notable in the late phase. Again, the wreath-like distribution with greatest density near the optic nerve is visible. 30-2 Humphrey Visual Field correlated with the patient’s history of an enlarged blind spot in the right eye. Each study was performed in the left eye as well, but all were unremarkable. Additional workup including CBC, CMP, ESR, CRP, FTA-ABS, ACE, lysozyme, quanterferon gold, and CXR were within normal limits, making MEWDS the presumptive diagnosis. On follow up ten days later, the patient reported improved blurriness and resolution of her scotoma without treatment, and OD visual acuity improved to 20/70 -2.
References:
- Raven ML, Ringeisen AL, Yonekawa Y, Stem MS, Faia LJ, Gottlieb JL. Multi-modal imaging and anatomic classification of the white dot syndromes. Int J Retina Vitr. 2017;3:12. doi:10.1186/s40942-017-0069-8.
- McCannel, Colin. 2017-2018 Basic and Clinical Science Course: Retina and Vitreous: American Academy of Ophthalmology; 2017.
- Bagheri N, Wajda BN, Calvo CM, Durrani AK, Friedberg MA, Rapuano CJ. The Wills Eye Manual: Office and Emergency Room Diagnosis and Treatment of Eye Disease. Seventh edition. Philadelphia: LWW; 2016.
Identifier: Moran_CORE_24094
Faculty Approval by: Griffin Jardine, MD
Copyright statement: Copyright J. Erik Kulenkamp, ©2017. Please see terms of use page for more information.
Choroidal Coloboma Image Report
Home / Pediatric Ophthalmology and Strabismus / Disorders of the Retina and Vitreous
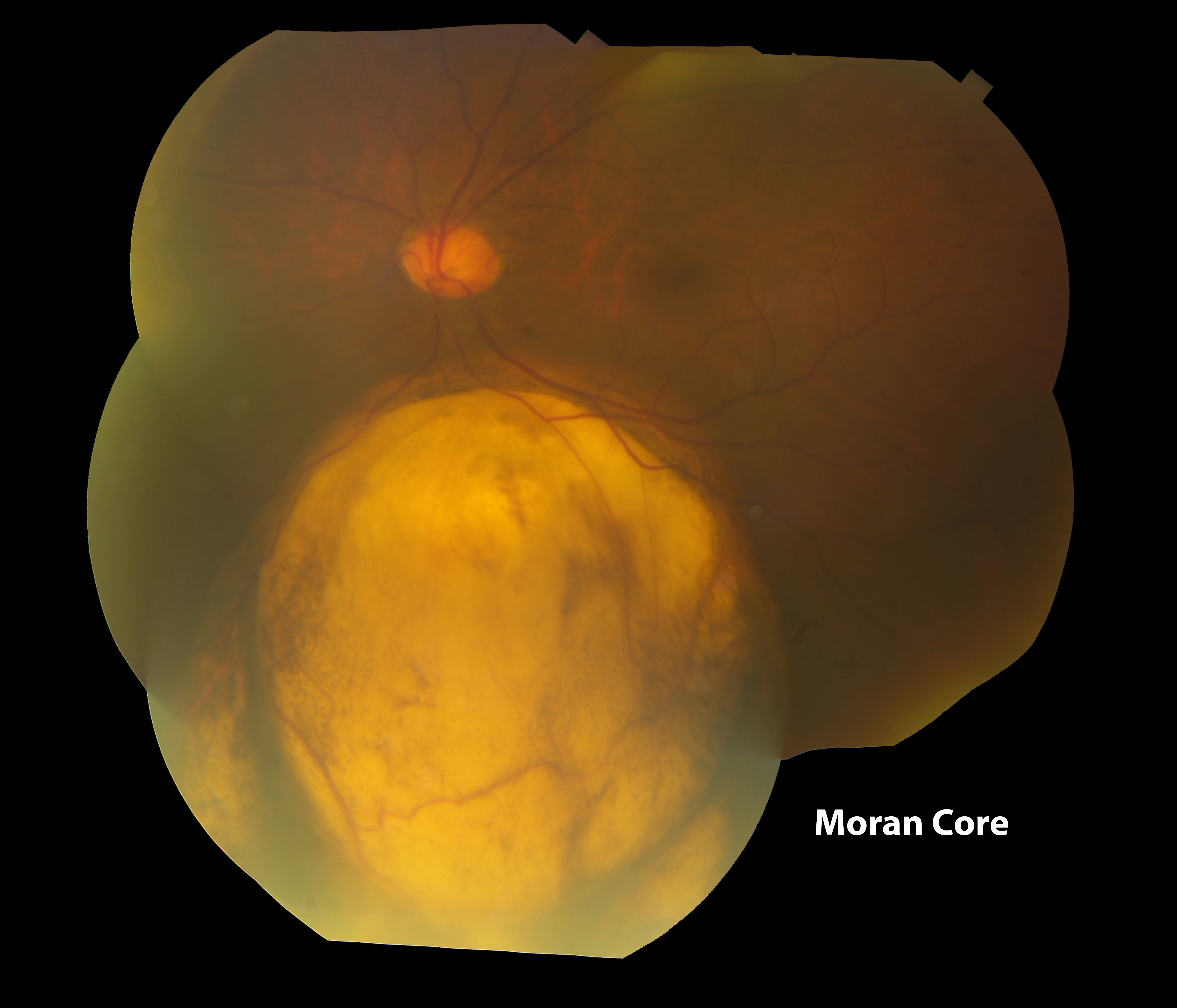
Figure 1. Montage color fundus photograph of the left eye demonstrating an anomalous optic nerve and a choroidal coloboma
Title: Choroidal Coloboma Image Report
Author(s): Eileen S. Hwang, MD, PhD; Akbar Shakoor, MD
Photographer:
Date: 05/18/2015
Image: Figure 1. Montage color fundus photograph of the left eye demonstrating an anomalous optic nerve and a choroidal coloboma
Keywords/Main Subjects: Choroidal Coloboma
Diagnosis: Choroidal Coloboma
Brief Description: The patient is a 65 year old female who presented for an abnormality found on a diabetic screening examination. She reported that she had a history of a congenital abnormality of her left eye and had poor vision in that eye since she was young. On examination, her visual acuity in the left eye with correction was 20/200. She had an iris coloboma from 6:00 to 8:00. Her left optic nerve had an anomalous appearance. She had a large well-demarcated, hypopigmented, excavated area of her retina inferior to the optic disc in the left eye.
Choroidal coloboma is a rare developmental abnormality that is caused by problems with closure of the embryonic fissure. In the area of the coloboma, the choroid and the outer layers of the retina are missing. On the surface of the coloboma, there is a thin intercalary membrane consisting of inner layers of the retina. The intercalary membrane is prone to breaks, and patients with choroidal colobomas may have up to a 40% risk of retinal detachment (Gopal, 1998). Detachments are most likely to occur in the second and third decades of life. In the absence of a retinal break or detachment, a choroidal coloboma can be observed. When intervention is required, diode laser demarcation of the coloboma is preferred over argon laser since diode laser causes less damage to the nerve fiber layer. To tamponade retinal detachments in patients with choroidal colobomas, silicone oil has been used with success (Schubert, 2005).
References: Schubert HD. Structural organization of choroidal colobomas of young and adult patients and mechanism of retinal detachment. Trans Am Ophthalmol Soc. 2005;103:457-72.
Gopal L, Badrinath SS, Sharma T, Parikh SN, Shanmugam MS, Bhende PS, Agrawal R, Deshpande DA. Surgical management of retinal detachments related to coloboma of the choroid. Ophthalmology. 1998 May;105(5):804-9.
Relevant links: Ort, Victoria, and David Howard. Development of the Eye. Retrieved 9 June 2015. http://education.med.nyu.edu/courses/macrostructure/lectures/lec_images/eye.html
Series: Moran Eye Center Image Report
Identifier: Moran_CORE_301
Copyright statement: ©2015. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/