


Ultrasound of a Patient with Terson Syndrome Secondary to a Gunshot Wound
Retina and Vitreous / Other Retinal Vascular Diseases
Title: Ultrasound of a Patient with Terson Syndrome Secondary to a Gunshot Wound
Author: Mark Parsons, MS4
Photographer: Roger Harrie, MD
Date: July 2, 2024
Secondary CORE Category: Retina and Vitreous / Posterior Segment Manifestations of Trauma; Ultrasound
Keywords/Main Subjects: Terson syndrome, intracranial hemorrhage, intraocular hemorrhage, trauma, ultrasound
Diagnosis: Terson Syndrome
Description of Case:
The images shown are from 23 year-old male who originally presented to the emergency department following a gunshot wound to the head. At the time, he underwent emergent hemicraniectomy for resection of a right temporal lobe hematoma, with concomitant subarachnoid and subdural hemorrhages noted on imaging. He experienced remarkable cognitive recovery and was discharged to a rehabilitation facility 18 days after presentation. Upon arrival at the new facility, ophthalmology was consulted for evaluation of visual complaints. The patient reported limited vision, ophthalmoplegia, and ptosis in the right eye (OD) that had been present since first awakening in the hospital. Evaluation was concerning for orbital apex syndrome and Terson syndrome, and B scan (as shown above) revealed vitreous hemorrhage in addition to focal subretinal, subhyaloid, or sub-internal limiting membrane (sub-ILM) hemorrhage. Vitrectomy with internal limiting membrane (ILM) peel was performed OD for removal of vitreous hemorrhage and what proved to be sub-ILM hemorrhage. At postoperative day 1, visual acuity (VA) was counting fingers at two feet. VA improved to 20/40 by postoperative day 9, and to 20/20 by postoperative week 6.
Defined most broadly, Terson syndrome is any intraocular hemorrhage in the setting of intracranial hemorrhage or an acute increase in intracranial pressure. The pathogenesis is not fully agreed upon, but a leading proposed mechanism is that rapid increase in intracranial pressure leads to an efflux of cerebrospinal fluid down the optic nerve sheath, compressing the central retinal vein and causing rupture of small retinal vessels.
Symptoms of Terson syndrome vary, with some patients being asymptomatic and others reporting loss of vision. Additionally, some may be unable to communicate their symptoms due to neurologic compromise. Fundoscopy may reveal varying forms of intraocular hemorrhage, including vitreous, subhyaloid, sub-ILM, intraretinal, or subretinal hemorrhages, either unilaterally or bilaterally. A classic “double ring sign” is sometimes present due to the presence of overlying subhyaloid and sub-ILM hemorrhages. If visualization is obscured or mydriasis is contraindicated, ultrasound or CT may be useful for diagnosis. Ultrasound is particularly helpful in cases of vitreous or preretinal hemorrhage. Most cases of Terson syndrome develop soon after the intracranial insult, but delayed presentations have been described. From an ophthalmic perspective, Terson syndrome may be managed via observation or vitrectomy. In cases of sub-ILM hemorrhage, an ILM peel may also be performed intraoperatively.

Figure 1. B-scan ultrasound of the affected eye. Vitreous hemorrhage can be seen over the posterior pole, with a second underlying hemorrhage, labeled SRF in the image. This was found to represent a sub-ILM hemorrhage intraoperatively.
Summary of the Case: A 23 year-old male was evaluated for Terson syndrome after being hospitalized for intracranial hemorrhages secondary to a gunshot wound to the head. B-scan revealed vitreous hemorrhage in the right eye with an additional second hemorrhage, which proved to be a sub-ILM hemorrhage. He underwent vitrectomy with ILM peel, and VA improved from counting fingers to 20/20 by postoperative week 6.
References:
- Ko F, Knox DL. The ocular pathology of Terson’s syndrome. Ophthalmology. 2010;117(7):1423-9.e2. doi:10.1016/j.ophtha.2009.11.028
- Reale C, Brigandì A, Gorgoglione N, Laganà A, Girlanda P. Terson’s syndrome. Practical Neurology. 2020;20(2):163-164. doi:10.1136/practneurol-2019-002326
- Stevanovic M, Eliott D. Terson Syndrome: A Review of the Literature. International Ophthalmology Clinics. 2024;64(2):89. doi:10.1097/IIO.0000000000000502
- Terson Syndrome: Don’t Let It Go Unrecognized. American Academy of Ophthalmology. Published October 31, 2018. Accessed June 30, 2024. https://www.aao.org/eyenet/article/terson-syndrome-dont-let-it-go-unrecognized
Faculty Approval by: Akbar Shakoor, MD
Copyright statement: Copyright Mark Parsons, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
A Case of Fulminant Serratia Marcescens Panophthalmitis after Penetrating Keratoplasty
Title: A Case of Fulminant Serratia Marcescens Panophthalmitis after Penetrating Keratoplasty
Authors: Meghan Sharma, MD, MPH1; Nour Bundogji, MD2; Nadim S. Azar, MD1; Amy Lin, MD1; Austin Nakatsuka, MD1
Affiliations:
1 John A. Moran Eye Center, University of Utah, Salt Lake City, UT, USA
2 John H. Stroger Jr. Hospital of Cook County, Chicago, IL, USA
Date: 6/24/2024
Keywords/Main Subjects: Fulminant panophthalmitis; Serratia marcescens panophthalmitis; penetrating keratoplasty; Serratia marcescens
Diagnosis: Fulminant Panophthalmitis
Description of Case: This is the first reported case of fulminant Serratia marcescens panophthalmitis after penetrating keratoplasty (PK).
Images:
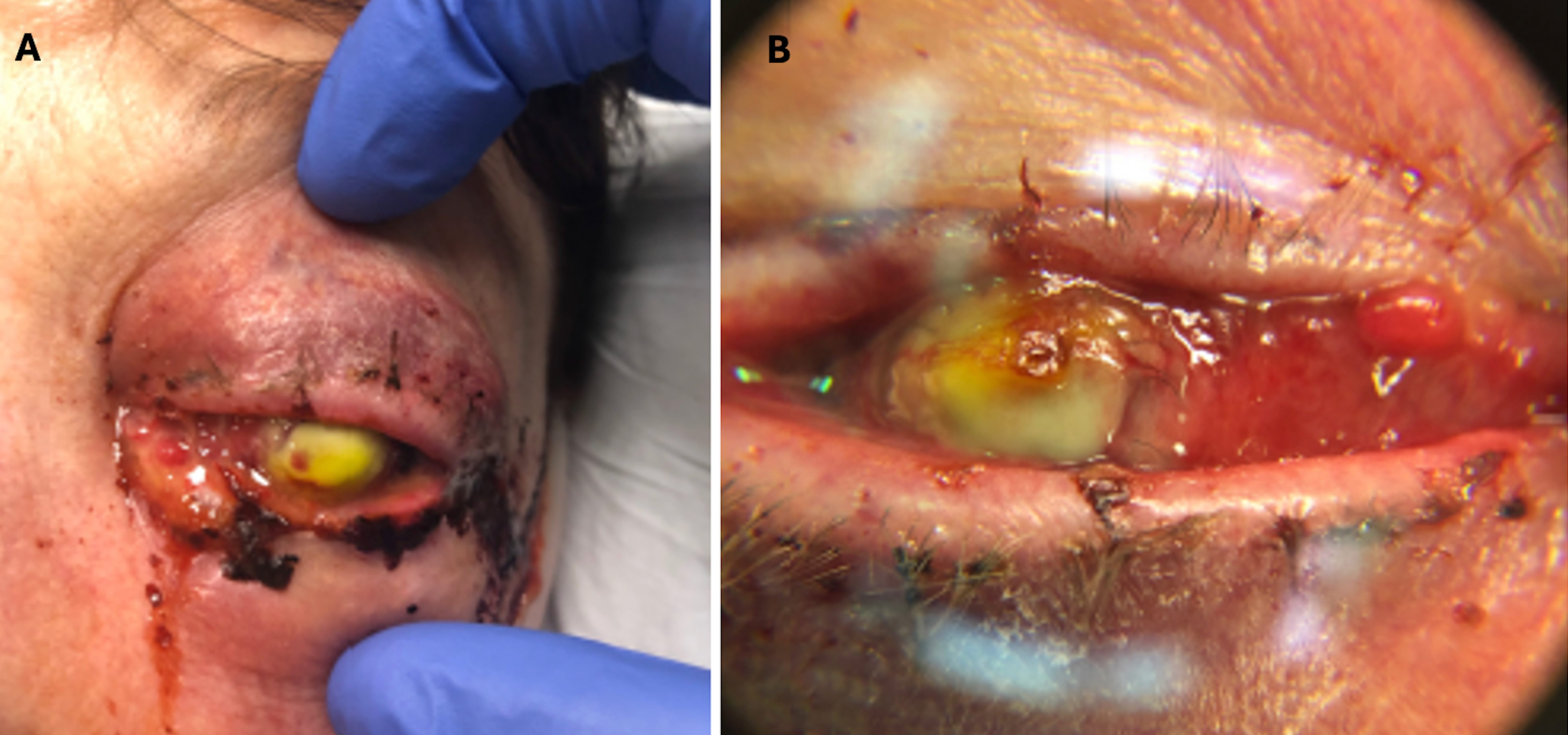
Figure 1
Figure 1A. External photograph of the patient’s left eye on post-operative day 2 following penetrating keratoplasty and secondary anterior chamber intraocular lens showing significant periorbital swelling, ecchymosis, and erythema as well purulent discharge and complete opacification of the cornea.
Figure 1B. Magnified photo of the patient’s left eye showing nasal chemosis, opacity of the PK graft, and diffuse, perforate corneal ulcer. PK graft appears to be in place and thickened and elevated nasally.
Note: The notable yellow tinge is fluorescein dye with complete uptake in the donor cornea.
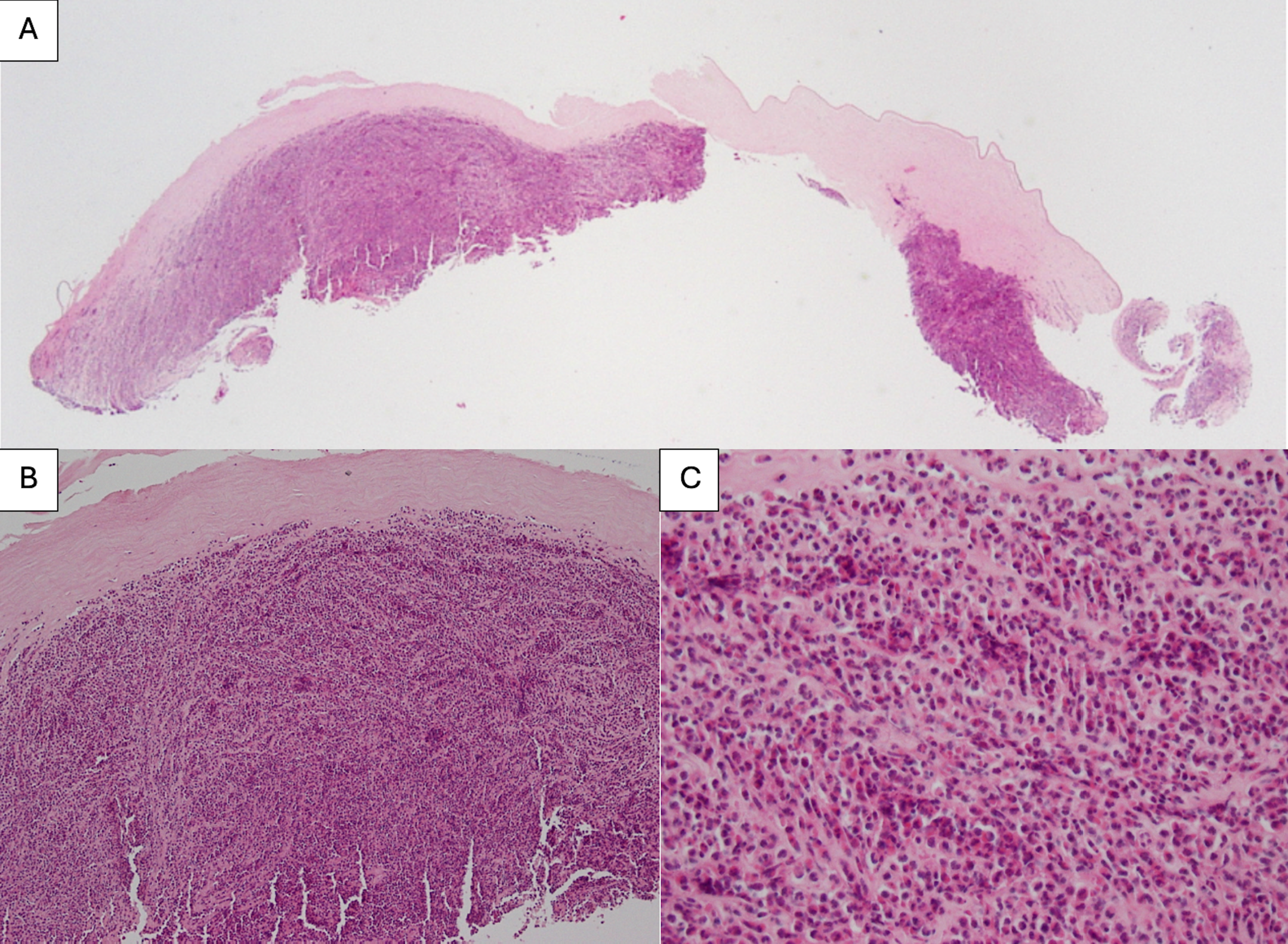
Figure 2
Figure 2A. Histopathology consistent with an intense corneal ulcer at 20X magnification. The epithelium, Bowman’s, and endothelial cell layers are completely absent. Posteriorly, there are fragments of Descemet’s membrane showing folding and wrinkling.
Figure 2B. Histopathology at 200X magnification. The stroma shows a marked inflammatory cell reaction that is almost full thickness.
Figure 2C. Histopathology at 400X magnification. There is mixed inflammation consisting mostly of acute inflammatory cells with polymorphonuclear neutrophils, rare eosinophils, and occasional lymphocytes and plasma cells.
ABSTRACT
Purpose:
To report the first case of fulminant Serratia marcescens panophthalmitis after penetrating keratoplasty (PK).
Methods:
This is a report of a patient who developed fulminant panophthalmitis shortly after undergoing a PK with anterior chamber (AC) intraocular lens (IOL) placement. Slit-lamp examination as well as B-scan ultrasound (B scan) and orbital CT of the left eye (OS) were performed to further evaluate the patient. Tissue culture and histopathologic examination of the corneal specimen were completed to confirm the diagnosis.
Results:
A 78-year-old pseudophakic female presented with two days of increasing pain, swelling, and purulent discharge after uneventful PK and secondary AC-IOL placement OS. Examination was notable for light proception without projection, elevated intraocular pressure of 48 mmHg, and a perforated corneal ulcer. B scan demonstrated diffuse vitreous opacities and membranes. Orbital CT demonstrated proptosis and high attenuation material within the left globe. Canthotomy, vitreous sampling, and antibiotic injections were performed. Corneal tissue cultures grew S. marcescens. Therapeutic PK was performed, but after rapid decompensation, the eye was eviscerated.
Conclusion:
This is the only reported case of fulminant S. marcescens panophthalmitis after penetrating keratoplasty. S. marcescens panophthalmitis is an aggressive and rapidly progressive infection with poor visual outcomes despite appropriate intravitreal and systemic antibiotic therapy.
INTRODUCTION
Endophthalmitis, defined as inflammation of the inner structures of the eye with exudation in the vitreous cavity, is a rare but serious complication that may follow penetrating keratoplasty (PK). [1, 2] Endophthalmitis may progress to panophthalmitis, which involves the sclera and Tenon’s capsule, if the infection is not controlled promptly with antibiotics. [3] Progression of endophthalmitis to panophthalmitis following PK is extremely rare, with a 2018 study reporting that only 3.03% of panophthalmitis cases are post-PK. [2] Moreover, studies have often attributed post-PK endophthalmitis with gram-positive organisms as the most common cause of infection. [4] Only one case to our knowledge has been reported on endophthalmitis following PK due to Serratia marcescens, a gram-negative bacterium. [5] In this study, we report the first known case of S. marcescens fulminant panophthalmitis following PK.
MATERIALS AND METHODS
A search using the PubMed database was performed to identify relevant literature regarding post-PK endophthalmitis, post-PK panophthalmitis, S. marcescens endophthalmitis, and S. marcescens panophthalmitis. We report the first known case of fulminant S. marcescens panophthalmitis. We discuss the patient’s slit-lamp examination, B-scan ultrasound, and orbital computed tomography (CT) of the left eye (OS). Tissue culture and histopathologic examination of the corneal specimen confirming the diagnosis are also included in the report.
RESULTS
A 78-year-old pseudophakic female with a history of bilateral (OU) keratoconus status post PK and Descemet stripping endothelial keratoplasty presented with intraocular lens (IOL) dislocation and a seidel positive PK after slipping on wet leaves and falling on the ground. Routine cultures were not performed on the donor PK at the time. Eight months later, she underwent a secondary anterior chamber IOL procedure and repeat PK of the left eye (OS); however, she began experiencing increasing OS pain two days after the procedure. On post-operative day three, she presented to the emergency department with OS pain, redness, swelling, and purulent discharge (Figure 1). Ophthalmic exam demonstrated light perception (LP) visual acuity in the affected eye with an intraocular pressure (IOP) of 48. Slit lamp exam was notable for a diffuse, perforate corneal ulcer and no view to the anterior chamber or posterior pole but bloody vitreous. A B-scan ultrasound was performed, showing diffuse vitreous opacities and membranes. Orbital CT showed proptosis with high attenuation material within the left globe, likely representing hemorrhage with superimposed infection. Lateral canthotomy and cantholysis were performed in addition to a tap and injection of vancomycin with ceftazotide. Aerobic, viral, fungal, and anaerobic cultures were taken from the cornea during the tap. Following the procedure, the patient was empirically treated for panophthalmitis with IV vancomycin and piperacillin-tazobactam while being maintained on fortified antibiotics in the left eye, including vancomycin and tobramycin every one hour. Cultures of the corneal tissue grew 3+ gram-negative rods which were noted to be S. marcescens, sensitive to cefepime, ceftazidime, tobramycin, and fluoroquinolones.
The patient was diagnosed with fulminant S. marcescens panophthalmitis of the left eye, as evident from the ophthalmic exam, orbital CT, and bacterial cultures. Treatment was modified to include moxifloxacin 400 mg IV daily, cefepime 2g every 8 hours, and oral doxycycline to minimize corneal thinning. On post-operative day four, the patient underwent therapeutic PK, drainage of suprachoroidal hemorrhage, scleral patch graft, and repeat injections of the left eye. However, the patient continued to experience worsening pain despite treatment with oxycodone. The eye was firm to palpation with resistance to retropulsion, as intraocular and intraorbital pressure had increased. Slit lamp exam showed periorbital edema and ecchymoses with proptosis, disordered and atrophic conjunctiva with dried hemorrhage, a dark patch of likely necrotic sclera inferonasally that was seidel negative, and exposed sclera temporally and nasally. The PK graft was in place and seidel negative but was thickened and elevated nasally. Given the patient’s worsening pain, uncontrolled orbital pressure, and poor visual prognosis, evisceration OS was planned.
In addition to evisceration OS with an 18 mm silicone implant, the patient also underwent left temporary suture tarsorrhaphy and left lateral canthoplasty for fulminant S. marcescens panophthalmitis in a blind, painful eye. Subsequent histopathology (Figure 2) of the cornea was consistent with an intense corneal ulcer demonstrating complete absence of the epithelium, Bowman’s, and endothelial cell layers as well as an almost full thickness acute inflammatory cell reaction. Nine days after evisceration, the patient did not endorse any eye pain.
DISCUSSION
This is a rare case of a 78-year-old female with fulminant S. marcescens panophthalmitis following standard PK which resulted in evisceration of the eye. The literature regarding endophthalmitis or panophthalmitis following PK is limited. Of the literature reviewed, the most common pathogens in post-PK endophthalmitis are gram-positive bacteria. [4] One study analyzed 1,010 consecutive PKs and found three cases of bacterial endophthalmitis all caused by Streptococci with one case of Candida albicans endophthalmitis. [6] A 2003 review of 1,074 cases of endophthalmitis at Wills Eye Hospital identified ten cases of post-PK endophthalmitis that were due to gram-positive cocci (six Streptococcus species, three Staphylococcus species, and one identified on pathology specimen only) and only three cases were due to gram-negative organisms (Proteus mirabilis, Serratia marcescens, and one identified on pathology specimen only). The case of post-PK S. marcescens endophthalmitis in the Wills Eye Hospital study was resistant to ampicillin, bacitracin, and cefazolin and was only sensitive to ceftazidime. No specific information was found regarding treatment or prognosis in this patient. [5] This contrasts with our case of post-PK S. marcescens infection, which was sensitive to cefepime, ceftazidime, tobramycin, and fluoroquinolones. While the previous study details a case of S. marcescens endophthalmitis following PK, only three other cases of S. marcescens panophthalmitis have been found in literature. Unlike our case, these reported cases did not follow an ophthalmic surgery such as a PK, as the sources were endogenous. All patients in these cases reached LP vision or required evisceration or enucleation. [3, 7]
- marcescens is a gram-negative, opportunistic bacterium of the family Enterobacteriaceae and is the second most common pathogen among hospital-acquired ocular infections after Pseudomonas aeruginosa. [3] The species is typically found in water, soil, the gastrointestinal tract, and the urinary tract; however, our patient had no known predisposing factors for S. marcescens infection. [8] S. marcescens endophthalmitis may sometimes be observed as a pink hypopyon due to its distinctive red pigment, but this is not always seen, as in our patient. [8, 9] According to animal studies of S. marcescens ocular infection, the mechanism of action of S. marcescens appears to be a destructive process characterized by a hyperacute suppurative inflammatory response with liquefactive necrosis of ocular tissues thought to be mediated by the production of proteases. [10] S. marcescens endophthalmitis has been noted to have a poor prognosis. The largest reported series of patients with S. marcescens endophthalmitis noted that half (5/10) of patients had a final visual acuity of NLP or required evisceration, similar to the patient in this case report who required evisceration. [8]
Data on the treatment of S. marcescens endophthalmitis is limited. In the Endophthalmitis Vitrectomy Study, a multicenter randomized clinical trial examining systemic antibiotic treatment in the management of postoperative endophthalmitis, S. marcescens was not identified in any of the culture-positive isolates. [9] According to literature, empiric antibiotic treatment for post-PK endophthalmitis includes vancomycin to cover gram-positive bacteria and ceftazidime or gentamicin for gram-negative bacteria. [5] Fluoroquinolones have rapid bactericidal action by inhibiting bacterial DNA gyrase and topoisomerase IV and may be considered in the treatment of S. marcescens. In a 2007 in vitro study examining antibiotic effects and corneal epithelial toxicity of levofloxacin and moxifloxacin on human corneal epithelial cells, moxifloxacin was more effective for S. marcescens than levofloxacin; however, moxifloxacin seemed to exhibit more toxicity on human corneal epithelial cells at high concentrations and with long-term use. [11] S. marcescens may also demonstrate resistance to initial antibiotic therapy or require further therapy, as one study reported S. marcescens resistance to gentamicin as high as 90%. [9] In an institutional review of S. marcescens endophthalmitis cases at Bascom Palmer Eye Institute, 10% of patients demonstrated persistent growth of S. marcescens on repeat vitreous cultures, and several others had further clinical deterioration despite use of sensitive antibiotics. [9] Similarly, our patient in this case report was sensitive to several antibiotics while on moxifloxacin and cefepime; however, her pain continued to worsen with no improvement in vision, ultimately leading to evisceration. In conclusion, this case study emphasizes that S. marcescens panophthalmitis is an aggressive and rapidly progressive infection with poor visual outcomes despite appropriate intravitreal and systemic antibiotic therapy.
References:
- Chen JY, Jones MN, Srinivasan S, et al., Endophthalmitis after penetrating keratoplasty. Ophthalmology, 2015. 122(1): p. 25-30.
- Pappuru RR, Dave VP, Pathengay A, et al., Endophthalmitis Progressing to Panophthalmitis: Clinical Features, Demographic Profile, and Factors Predicting Outcome. Semin Ophthalmol, 2018. 33(5): p. 671-674.
- Guo HP, Wang TJ, Fulminant Serratia marcescens Panophthalmitis. Am J Med Sci, 2017. 353(4): p. e7.
- Alharbi SS, Alrajhi A, Alkahtani E, Endophthalmitis following keratoplasty: incidence, microbial profile, visual and structural outcomes. Ocul Immunol Inflamm, 2014. 22(3): p. 218-23.
- Kunimoto DY, Tasman W, Rapuano C, et al., Endophthalmitis after penetrating keratoplasty: microbiologic spectrum and susceptibility of isolates. Am J Ophthalmol, 2004. 137(2): p. 343-5.
- Kloess PM, Stulting RD, Waring GO, et al., Bacterial and fungal endophthalmitis after penetrating keratoplasty. Am J Ophthalmol, 1993. 115(3): p. 309-16.
- Breazzano MP, Jonna G, Nathan NR, et al., Endogenous Serratia marcescens panophthalmitis: A case series. Am J Ophthalmol Case Rep, 2019. 16: p. 100531.
- Goldenberg DT, Harinandan A, Walsh MK, et al., Serratia marcescens endophthalmitis after 20-gauge pars plana vitrectomy. Retin Cases Brief Rep, 2010. 4(2): p. 140-2.
- Bhikoo R, Blakiston M, Cunningham W, et al., Serratia Marcescens Endophthalmitis and Bacteraemia following Complicated Cataract Surgery. Ocul Immunol Inflamm, 2022. 30(4): p. 1020-1021.
- Sridhar J, Kuriyan AE, Flynn Jr. HW, et al., ENDOPHTHALMITIS CAUSED BY SERRATIA MARCESCENS: Clinical Features, Antibiotic Susceptibilities, and Treatment Outcomes. Retina, 2015. 35(6): p. 1095-100.
- Kim SY, Lim JA, Choi JS, et al., Comparison of antibiotic effect and corneal epithelial toxicity of levofloxacin and moxifloxacin in vitro. Cornea, 2007. 26(6): p. 720-5.
Faculty Approval by: Dr. Austin Nakatsuka
Copyright Statement: Copyright Meghan Sharma, Nour Bundogji, Nadim S. Azar, Amy Lin, Austin Nakatsuka, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
A Man with Bilateral Vision Loss
Title: A Man with Bilateral Vision Loss
Authors: Samuel W. Wilkinson1, MD, Ashley Polski , MD, Roger P. Harrie1, MD
Date: 6/18/24
Keywords/Main Subjects: Meningioma, disc edema, proptosis
Diagnosis: Atypical meningioma
Description of Case:
A 72-year-old male presented to the university ophthalmology triage clinic with a 3-week history of severe visual loss in his right eye and a recent mild dimming of vision in his left eye. During a prior workup with a retinal specialist, he was reportedly found to have right optic disc edema in the setting of normal erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). The retinologist suspected non-arteritic anterior ischemic optic neuropathy (NAION) in the right eye and told the patient there was no treatment. His past medical history included essential hypertension, cataract surgery, and rheumatoid arthritis treated with daily 400 mg of hydroxychloroquine for 13 years. Examination of his right eye found visual acuity of count fingers at 1 meter with a 3+ afferent pupil defect and moderate optic disc edema. Vision in the left eye was 20/50 with a normal appearing optic disc. Intraocular pressures were 15 mmHg in both eyes. He was a moderate myope and had 2 mm proptosis of his right eye with a mild abduction deficit. A 24-2 Humphrey visual field test showed black-out on the right and moderate generalized depression on the left.
The patient was referred to the emergency department for a brain and orbital MRI scan, which showed a 5.2 x 3.4 x 4.5 cm mass centered in the sella, most compatible with an invasive pituitary macroadenoma (Figures 1 and 2). The mass invaded the skull base and extended into the right more than the left optic canals and into the right posterior extracortical space at the orbital apex. It also displaced the right more than the left optic nerve cisternal and canalicular segments. There were no findings of cavernous sinus invasion. The patient underwent a combined neurosurgery and otolaryngology procedure with debulking of the tumor. The pathology report was consistent with a grade II atypical meningothelial meningioma.
Discussion:
Meningiomas are the most common primary central nervous system tumor which comprise over 50% of benign tumors in the brain and spinal cord. The average age at diagnosis is 66 years old and they occur in women about twice as often as men. They tend to be more malignant when they occur in men and even more so in children.1 They arise from meningeal arachnoid cells and up to 2% of autopsies find meningiomas that were unknown to the patient during life because they were asymptomatic. They are categorized histologically into WHO grades I, II, and III based on increasing malignancy.2 The ten year survival rates are 84% for grade I, 53% for grade II, and 0% for grade III. The patient described in this report had a Grade II atypical meningioma with a 5 year recurrence rate of 50 to 55%. Treatment includes observation for asymptomatic tumors, surgical resection, and radiation.3
Patients such as the one in our case with optic disc edema and bilateral vision loss must be evaluated for orbital and intracranial etiologies, particularly when proptosis is present. The initial impression of NAION in the patient’s right eye was inconsistent with subsequent decreased vision in the left eye with a normal appearance of the optic disc. Second eye involvement in NAION occurs in 15 to 17% of patients over five years, but the exam would usually demonstrate sectoral disc edema and altitudinal visual field loss.4 In some cases, sequential vision loss occurs from an intracranial mass causing optic atrophy in the ipsilateral eye and papilledema in the contralateral eye—a condition known as Foster-Kennedy syndrome.5 The patient in this report did not technically fit the criteria for Foster-Kennedy syndrome with papilledema in only one eye and a normal appearing optic disc in the other, but he could have potentially manifested this finding if surgical intervention had not occurred.
A temporal artery biopsy to evaluate for giant cell arteritis (GCA) should be considered in elderly patients with visual loss in one eye and impending loss in the opposite eye, particularly if there are signs of vascular compromise to the optic disc and/or retina or other manifestations of systemic inflammation.6 Extraocular motility deficits can also, in rare cases, occur in the setting of GCA.7 Our patient had normal inflammatory markers (ESR and CRP) and denied symptoms such as tenderness in the temples and jaw claudication. Additionally, the finding of proptosis would be atypical for GCA and points more towards a mass with orbital involvement.
In patients with proptosis or extraocular motility abnormalities for whom an intracranial/orbital mass has been ruled out, a thyroid antibody panel would be a reasonable next step in the work-up. Patients with thyroid eye disease can, in rare cases, experience vision loss and optic disc edema due to optic nerve compression. More common clinical findings of thyroid eye disease include eyelid retraction, extraocular motility restriction, and conjunctival injection.8 A macular OCT to analyze the ellipsoid zone is also a reasonable consideration given his 13 year history of hydroxychloroquine use that could cause a maculopathy.9 A macular OCT is also helpful in ruling out other retinal pathology such as age-related macular degeneration which is a leading cause of vision loss in older adults.10 However, our patient did not have any findings of retinal pathology on dilated fundus exam.
Patient Outcome:
He had his pituitary mass resected, which found a WHO grade 2 atypical meningioma. During a follow up visit in neuro-ophthalmology clinic two months after resection of his tumor, his best-corrected vision improved from count fingers at 1 meter to 5/200 in his right eye and from 20/50 to 20/20 in his left eye. His right optic nerve was pale without edema and his left optic nerve was normal. Goldmann visual field testing found a central island of vision with marked peripheral constriction on the right and a small arcuate defect on the left.
Images or video:
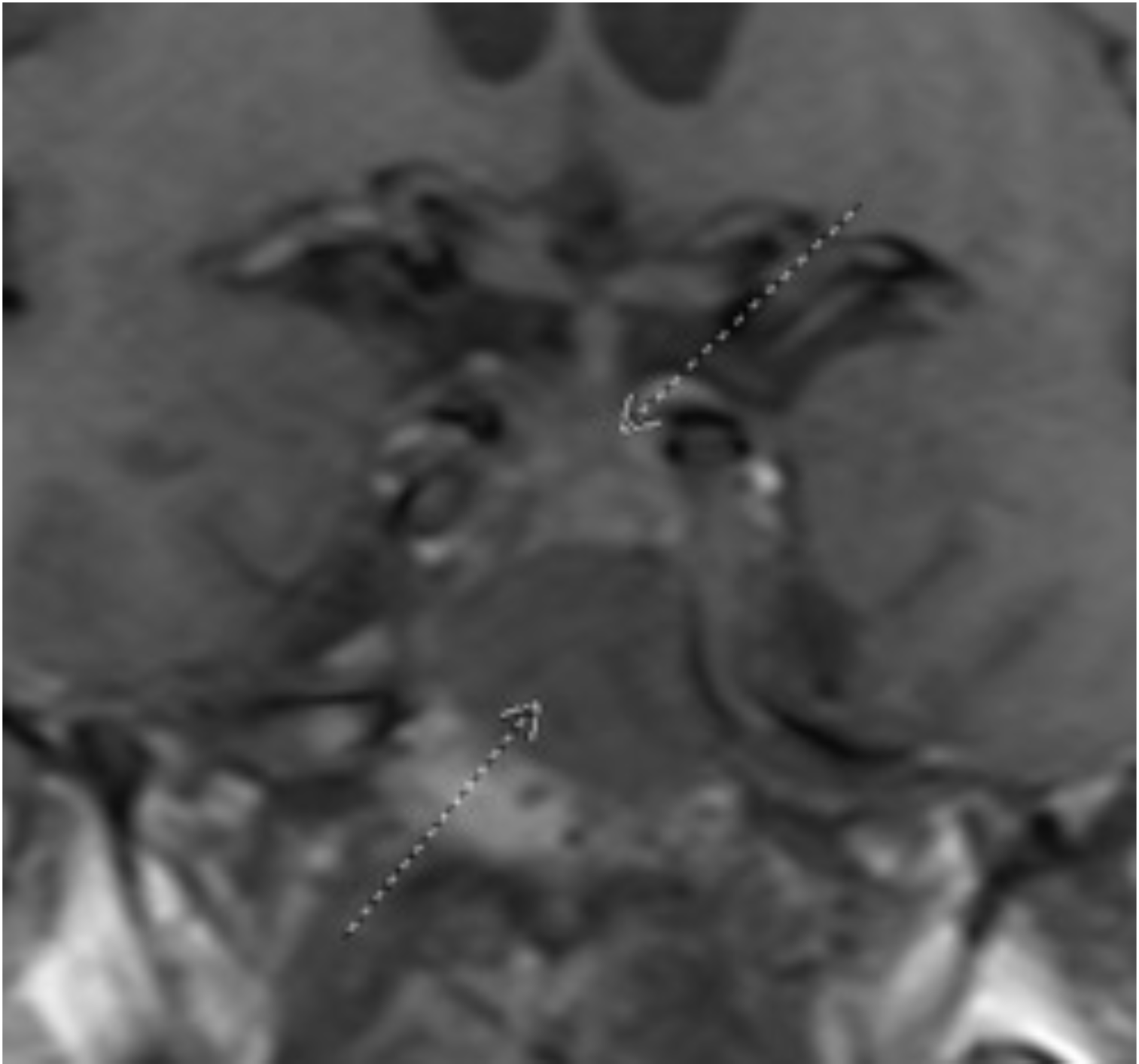
Figure 1: Coronal T1 view of a skull-based mass (inferior arrow) lying inferior to the optic chiasm (superior arrow)

Figure 2: Axial T1, post-contrast view of a skull-based mass located in the sella
Summary of the Case: A 72-year-old man referred with the diagnosis of NAION presented with severe visual loss in his right eye and recent mild dimming of vision in his left eye. Visual acuity OD was count fingers with moderate optic disc edema. Vision in the left eye was 20/50 with a normal optic disc. He had a 2 mm proptosis of his right eye with a mild abduction deficit. An MRI scan showed a large mass centered in the sella that compressed the right optic nerve. Pathology from surgical debulking of the tumor found a grade II atypical meningothelial meningioma.
References:
- Elefante A, Russo C, di Stasi M, Vola E, Ugga L, Tortora F, et al. Neuroimaging in meningiomas: old tips and new tricks. Mini-invasive Surg(2021) 5:7. doi: 10.20517/2574-1225.2020.102
- Buerki RA, Horbinski CM, Kruser T, Horowitz PM, James CD, Lukas RV. An overview of meningiomas. Future Oncol(2018) 14:2161–77. doi: 10.2217/fon-2018-0006
- Ogasawara C, Philbrick BD, Adamson DC. Meningioma: A Review of Epidemiology, Pathology, Diagnosis, Treatment, and Future Directions. Biomedicines2021, 9(3), 319; https://doi.org/10.3390/biomedicines9030319
- Newman NJ, Scherer R, Langenberg P, et al. The fellow eye in NAION: report from the ischemic optic neuropathy decompression trial follow-up study. Am J Ophthalmol. Sep 2002;134(3):317-28. doi:10.1016/s0002-9394(02)01639-2
- Sanders MD. The Foster Kennedy sign. Proc R Soc Med. Jun 1972;65(6):520-1.
- Vodopivec I, Rizzo JF. Ophthalmic manifestations of giant cell arteritis. Rheumatology (Oxford). Feb 01 2018;57(suppl_2):ii63-ii72. doi:10.1093/rheumatology/kex428
- Ross AG, Jivraj I, Rodriguez G, et al. Retrospective, Multicenter Comparison of the Clinical Presentation of Patients Presenting With Diplopia From Giant Cell Arteritis vs Other Causes. J Neuroophthalmol. Mar 2019;39(1):8-13. doi:10.1097/WNO.0000000000000656
- Weiler DL. Thyroid eye disease: a review. Clin Exp Optom. Jan 2017;100(1):20-25. doi:10.1111/cxo.12472
- Yusuf IH, Sharma S, Luqmani R, Downes SM. Hydroxychloroquine retinopathy. Eye (Lond). 2017;31(6):828-845. doi:10.1038/eye.2016.298
- Guymer RH, Campbell TG. Age-related macular degeneration. Lancet. Apr 29 2023 401(10386):1459-1472. doi:10.1016/S0140-6736(22)02609-5
Faculty Approval by: Roger Harrie, MD
Copyright statement: Sam Wilkinson, Ashley Polski, Roger P. Harrie ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Duane Syndrome
Home / Pediatric Ophthalmology and Strabismus / Special Forms of Strabismus
Title: Duane Syndrome
Author: Kerri McInnis-Smith, 4th year medical student, Mayo Clinic
Photographer: Dr. Marielle Young
Date: 7/14/2022
Keywords/Main Subjects: Strabismus, Duane Syndrome, Esotropia
Diagnosis: Duane Syndrome
Images or video:

Description of Case: Duane Syndrome, also known as Duane Retraction Syndrome (DRS), is a form of childhood strabismus characterized by globe retraction and palpebral fissure narrowing on attempted adduction.
Epidemiology: The prevalence of DRS in the general population is relatively low, affecting around 1/1000 individuals and accounting for <5% of all strabismus cases.1 Only one eye is involved in most cases, although up to 20% of affected patients may have bilateral involvement.2 For reasons that remain unclear, female patients are more commonly affected (60%) by DRS than male patients (40%) and the left eye is involved more often than the right eye.1,2 There is osme thought that the unilateral, left-sided, and female predominance could be due to the asymmetry in the thoracic anatomy and thrombosis risk factors.3
Pathophysiology: DRS is caused by abnormal (and sometimes absent) development of the abducens nerve (CN VI) between weeks 4-8 of embryological development. Occasionally, the lateral rectus muscle may receive aberrant innervation from the oculomotor nerve (CN III), contributing to concurrent horizontal recti action and subsequent globe retraction.4 Most cases (70%) of DRS are isolated to the below ocular findings, while around 30% are associated with additional ophthalmologic or systemic abnormalities. Some associated conditions include:2,5,6
- Okihiro’s syndrome: DRS + radial ray defects
- Wildervanck syndrome: DRS + Klippel-Feil anomaly + deafness
- Moebius syndrome: DRS + congenital facial palsy
- Hold-Oram syndrome: DRS + abnormalities of upper limbs and heart
- Morning Glory syndrome: abnormalities of optic disc
- Goldenhar syndrome: abnormalities eye, ear and spine
Risk factors: Although DRS occurs spontaneously in approximately 90% of isolated cases, around 10% of isolated cases are inherited.7 The only known risk factor for development of the condition is an affected biological parent. Various genes have been implicated in the inheritance of DRS, including mutations at locus 8q13 and in CHN1 on chromosome 2. Both autosomal dominant and autosomal recessive inheritance patterns have been demonstrated.2,5
Signs and symptoms:
- Complete or partial absence of abduction and/or adduction
- Retraction of globe on attempted adduction
- Narrowing of palpebral fissure on attempted adduction (induced ptosis)
- Abnormal head position (to compensate for duction deficit and maintain binocular single vision)
- Upshoots or downshoots (43% of cases2): affected eye deviates up/down with attempted adduction
- May occur secondary to mechanical effect (tight fibrotic muscles) or innervational anomalies2
Diagnosis: Diagnosis of DRS is typically made on clinical grounds alone, with additional imaging usually not necessary. Genetic testing may be pursued if familial inheritance is suspected.
Subtypes: Multiple criteria have been proposed to classify DRS according to clinical signs and symptoms. The most popular classification system, proposed by Huber et al, consists of 3 distinct subtypes:1
- Type I DRS (75-80%): mainly defective abduction, with normal or minimally defective adduction
- Esotropia in primary gaze compensatory head turn toward involved side
- Type II DRS (5-10%): mainly defective adduction, with normal or minimally defective abduction
- Exotropia in primary gaze compensatory head turn toward uninvolved side
- Type III DRS (10-20%): defective in both abduction and adduction
Individuals can also be sub-grouped according to their deviation in primary position, including esotropic DRS (eso-DRS), exotropic DRS (exo-DRS), and orthotropic DRS (exo-DRS).1 These two systems of classifying DRS can be helpful in the decision of whether to manage conservatively or surgically.
Differential diagnosis:5,7
- Abducens nerve palsy
- Congenital esotropia
- Brown Syndrome
- Marcus Gunn Jaw Winking Syndrome
- One of the associated systemic conditions mentioned above (Okihiro’s syndrome, Goldenhar syndrome, Wildervanck syndrome, Moebius syndrome, Holt-Oram syndrome, Morning Glory syndrome)
Management:
- Non-surgical: Not all individuals with DRS require surgical intervention. Conservative measures, such as observation, refractive correction, or prism glasses to improve abnormal head position are often sufficient to manage symptoms. Young patients should undergo repeat ophthalmology exams to assess for amblyopia. However, once a patient is not at a significant risk of developing amblyopia (around age 10), exams can occur less frequently.2
- Surgical: A subset of patients with DRS (estimated around 41%1) will progress to requiring surgical intervention. There are four generally accepted indications for which extraocular muscle surgery should be considered:1
- Significant abnormal head posture
- Significant deviation in primary position
- Severely abnormal eyelid position (retraction and narrowing of palpebral fissure)
- Significant upshoot or downshoot during adduction
The exact surgical approach is dependent on the patient’s symptoms, deviation in primary position, and specific duction deficits, but often consists of medial or lateral rectus recession and/or transposition of one or two vertical rectus muscles. Globe retraction can be improved via recessions of the co-contracting horizontal recti muscles.1
Potential complications: Although isolated DRS is not associated with severe complications, up to 10% of patients may develop amblyopia, especially without regular ophthalmologic exams.1
References:
- Gaballah KA. Treatment modalities in Duane’s Retraction Syndrome. Int J Ophthalmol. 2020 Feb 18;13(2):278–83.
- Kekunnaya R, Negalur M. Duane retraction syndrome: causes, effects and management strategies. Clin Ophthalmol. 2017 Oct;Volume 11:1917–30.
- Parsa CF, Robert MP. Thromboembolism and Congenital Malformations: From Duane Syndrome to Thalidomide Embryopathy. JAMA Ophthalmol. 2013 Apr 1;131(4):439.
- Hoyt W, Nachtigäller H. Anomalies of ocular motor nerves: Neuroanatomic correlates of paradoxical innervation in Duane’s syndrome and related congenital ocular motor disorders. Am J Ophthalmol. 1965 Sep;60(3):443–8.
- Graeber CP, Hunter DG, Engle EC. The Genetic Basis of Incomitant Strabismus: Consolidation of the Current Knowledge of the Genetic Foundations of Disease. Semin Ophthalmol. 2013 Sep;28(5–6):427–37.
- Kirkham TH. Duane’s syndrome and familial perceptive deafness. Br J Ophthalmol. 1969 May 1;53(5):335–9.
- Gaur N, Sharma P. Management of Duane retraction syndrome: A simplified approach. Indian J Ophthalmol. 2019;67(1):16.
Faculty Approval by: Marielle Young, MD
Identifier: Moran_CORE_127212
Copyright Kerri McInnis-Smith, ©2024. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Fundus Photography, Fluorescein Angiography and Optical Coherence Tomography of Bilateral Exudative Detachments in a Pediatric Patient
Home / Pediatric Ophthalmology and Strabismus / Disorders of the Retina and Vitreous
Title: Fundus Photography, Fluorescein Angiography and Optical Coherence Tomography of Bilateral Exudative Detachments in a Pediatric Patient
Author: Olaoluwa Omotowa, MPH, Nnana Amakiri, MD, Marcus Altman, MD, Theresa Long, MD
Keywords/Main Subjects: Bilateral Exudative Retinal Detachments
Diagnosis: Bilateral Exudative Retinal Detachments
-

- Figure 1: Optical Coherence Tomography of Right Eye demonstrating inferior serous retinal detachment sparing the macula with optic disc edema.
-
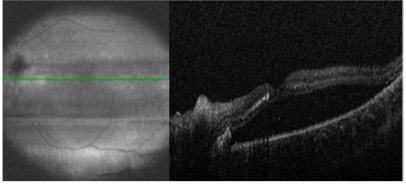
- Figure 2: Optical Coherence Tomography of Left Eye demonstrating serous retinal detachment with macular involvement and optic disc edema.
-

- Figure 3: Color Fundus demonstrating Bilateral Serous Retinal Detachments with peripapillary cotton wool spots and full detachment of the macula OS. Elschnig spots in periphery OU.
-

- Figure 4: Fluorescein Angiography demonstrating patchy choroidal filling with scattered, nonspecific, hypofluorescent lesions with late hyperfluorescent lesions OU.
Description of Case:
A 4-year-old girl with a history of chronic hypertension, albinism, hypothyroidism, and complicated delivery requiring an extended NICU stay was referred to our facility with progressive ataxia, facial weakness, and loss of appetite. She was afebrile without new rashes or constitutional symptoms. The patient’s medical history included a seizure during NICU stay and a diagnosis of Bell’s Palsy at age 1. Additionally, she had a history of being diffusely edematous and hypertensive while in the NICU, which led to treatment with anti-hypertensive medications. At home her systolic blood pressures were reportedly in the 130-140s. Initial CT at an outside hospital revealed ventriculomegaly and brainstem glioma prompting urgent referral to our facility and neurosurgical consultation with administration of 4mg dexamethasone IV.
During her current presentation, she developed bilateral facial weakness, including ptosis related to Bell’s Palsy, along with her progressive ataxic gait, right ear pain, decreased appetite, and occasional emesis. On arrival, she had age-appropriate vital signs (94/58) and normal mental status.
Her neurological status deteriorated, necessitating an MRI scan. While lying flat in the MRI scanner, she became hypertensive with SBP in 180s. The scan revealed worsening ventriculomegaly, longitudinally extensive transverse myelitis, bilateral retinal hemorrhages, and nonenhancing T2/FLAIR signal in various brain regions. She received a total of 18 mg labetalol and ultimately required nicardipine drip 1.5 mcg/kg/hr normalizing her blood pressure to 138/100. She was intubated in MRI and taken for emergent EVD placement with neurosurgery. She tolerated the procedure well without complications and was subsequently weaned off her nicardipine drip. Given the ocular findings on imaging, ophthalmology was promptly consulted.
Ophthalmic examination revealed no light perception in both eyes both of which were soft to palpation. She had mild esophoria and no afferent pupillary defect though was on miotics. Her fundus examination revealed bilateral exudative retinal detachments, tortuous vessels, and diffuse serous detachments with a tigroid appearance and diffuse creamy infiltrates. Further evaluation, including ocular ultrasonography and fluorescein angiography, was performed to ascertain the underlying cause.
A comprehensive workup, including laboratory investigations, lumbar puncture, whole-genome sequencing, and whole body imaging was initiated to identify the etiology of the bilateral exudative retinal detachments. Laboratory results indicated AQP4-IgG, Adams13, MOG Ab IgG, D-dimer, Haptoglobin, RPR, TSH, C3, C4 CRP, and ESR were within normal limits. RCIGM revealed no genomic variants. Anti-CFH Autoantibody, HSV IgG/IgM, B Burgdorferi IgM & IgG, Quant Gold, Cat Scratch IgG & IgM, ANCA, HIV, Treponema, meningitis/encephalitis panel, oligoclonal bands, CSF flow and cytology were negative. LDH (740), platelets (316), IgG (333), IgG synthesis (14.5), and Von Willebrand Ag (150%) were all elevated. The patient’s clinical and diagnostic findings led to a diagnosis of hypertensive chorioretinopathy as the most likely cause.
Patient was scheduled for close follow up 2 weeks after her initial diagnosis. After not making it to this appointment she was rescheduled with her hometown ophthalmologist. These visits demonstrated gradual resolution of subretinal fluid and exudates. Serial optical coherence tomography scans and dilated examinations demonstrated reattachment of the retina in both eyes, with the patient’s visual acuity improving, although some residual impairment remained due to macular involvement. She was 20/300 in her right eye and 20/200 in her left.
Summary of the Case:
- Bilateral exudative retinal detachments in pediatric patients present diagnostic challenges due to their rarity and various potential etiologies.
- The causes can be classified as inflammatory, infectious, or neoplastic, including conditions like familial exudative vitreoretinopathy, hemolytic uremic syndrome, choroidal lymphoma, hemangioma, and metastases.
- The presented case highlights the importance of considering rare causes, such as malignant hypertension, in patients with bilateral exudative retinal detachments. Prompt control of blood pressure played a crucial role in the resolution of fundus findings.
- This case emphasizes the significance of thorough evaluation and heightened awareness among eyecare providers when encountering pediatric patients with bilateral exudative retinal detachments. Timely diagnosis and management are crucial to prevent irreversible vision loss.
- Hypertensive chorioretinopathy should be considered as a possible etiology in cases of acute on chronic hypertension presenting with retinal detachments.
References:
- Yoshida, K., Hasegawa, D., Takusagawa, A., Kato, I., Ogawa, C., Echizen, N., … & Manabe, A. (2010). Bullous exudative retinal detachment due to infiltration of leukemic cells in a child with acute lymphoblastic leukemia. International journal of hematology, 92, 535-537.
- Rosecan, L. R., Laskin, O. L., Kalman, C. M., Haik, B. G., & Ellsworth, R. M. (1986). Antiviral therapy with ganciclovir for cytomegalovirus retinitis and bilateral exudative retinal detachments in an immunocompromised child. Ophthalmology, 93(11), 1401-1407.
- Navarrete, A., Jaouni, T., & Amer, R. (2023). Total exudative retinal detachment in a child with pars planitis-a challenging case with optimistic results. Journal of Ophthalmic Inflammation and Infection, 13(1), 1-4.
- Otuka, O. A. I., Eweputanna, L. I., Okoronkwo, N. C., & Kalu, A. (2021). Bilateral Exudative Retinal Detachment in a Young Patient with Chronic Renal Failure. International Medical Case Reports Journal, 139-144.
- Khaja, W. A., Pogrebniak, A. E., & Bolling, J. P. (2015). Combined orbital proptosis and exudative retinal detachment as initial manifestations of acute myeloid leukemia. Journal of American Association for Pediatric Ophthalmology and Strabismus, 19(5), 479-
- Shukla UV, Gupta A, Tripathy K. Exudative Retinal Detachment. 2023 Feb 22. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 36944005.
Faculty Approval by: Theresa Long, MD
Copyright statement: Olaoluwa Omotowa, ©2023. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Intraocular Pressure Fluctuation in a Patient with Pseudoexfoliation Glaucoma
Title: Intraocular Pressure Fluctuation in a Patient with Pseudoexfoliation Glaucoma
Authors: Tara Gallant, California Northstate University College of Medicine, MD Class of 2024; Barbara Wirostko, MD
Date: September 2023
Keywords/Main Subjects: Pseudoexfoliation Syndrome, Pseudoexfoliation Glaucoma, Intraocular Pressure Fluctuations, Home iCare
Introduction:
Pseudoexfoliation syndrome (XFS) results from abnormal fibrillar extracellular material accumulation in ocular tissues, including all structures of the anterior segment, the conjunctiva, and orbital structures.1 XFS is one of the most common causes of open-angle glaucoma. Pseudoexfoliation glaucoma (XFG) typically presents with a higher maximum and mean IOP at the time of diagnosis, as well as a wider range of IOP fluctuation, compared with primary open angle glaucoma (POAG). It is associated with a higher risk of progressive vision loss than POAG, more rapidly developing cataracts, phacodenesis, lens subluxation, and retinal vascular events.1,2 XFS manifestations are age-related and estimated to be present in 10-20% of the general population above the age of 60 depending upon the geographic location.2 Patients with XFS are thought to have a ten-fold higher risk of developing glaucoma than the general population.2 On dilated clinical exam, XFS can be identified by the presence of white deposits on the anterior lens surface and pupillary margins. Ultrasound biomicroscopy often reveals zonular weakness, a thickened lens, a narrow anterior chamber, and occludable angles.2 Systemic diseases with an increased incidence in patients with XFS include chronic obstructive pulmonary disease, inguinal hernias, pelvic organ prolapse, obstructive sleep apnea, and atrial fibrillation.3
Case Presentation:
Our patient is an 81-year-old female with severe stage capsular pseudoexfoliation glaucoma in both eyes. Five years ago, she underwent Phaco CyPASS surgeries in both eyes. Approximately two years ago, the patient had a XEN gel stent with mitomycin C/Ologen placed in the right eye. Her current ocular medications include Cosopt BID OU and Vyzulta QHS OU. Other ocular conditions include dry eye syndrome and meibomitis. She has a history of pelvic organ prolapse, and her mother was also diagnosed with glaucoma and pelvic organ prolapse.
At her most recent visit, visual acuity was 20/30 OD and 20/25 OS, with applanation tonometry readings of 8 OD and 12 OS. Slit lamp exam was notable for pseudoexfoliation material on the anterior aspect of the lens capsule with no transillumination defects bilaterally. She was also noted to have phacodenesis OD. Her right optic nerve had temporal pallor and a cup/disc ratio of 0.95, and her left optic nerve had an infratemporal notch with a cup/disc ratio of 0.6. Other exam findings included trace meibomian gland dysfunction OU, trace hordeolum OU, and well-centered posterior chamber intraocular lenses bilaterally. Optic nerve OCT scans demonstrated diffuse temporal thinning OD and infratemporal thinning OS, with no progression of glaucomatous damage over the past year and a half in either eye when compared to prior scans.
Two and a half years ago, the patient was assigned an iCare HOME to determine if she was experiencing intraocular pressure (IOP) fluctuations that could help explain the severe and progressive glaucomatous damage to her optic nerves, particularly in her right eye. A summary of the results is shown in Figure 1 below. The measurements taken revealed higher overall IOP in the right eye outside of normal clinic hours, as well as increased fluctuation in pressures when compared to the left eye. The highest and lowest measurements taken are shown in Table 1 below.

Figure 1: Summary of the results of iCare Home measurements taken three months prior to the glaucoma stent procedure OD. All the measurements were “excellent” in quality. Average IOP and variability of IOP was higher in the right eye, which had more glaucomatous damage compared to the left eye. Both average IOP and variability of IOP in OD were higher on these iCare measurements than on clinical measurements around the same time.

Table 1: Highest and lowest IOP measurements taken in each eye while the patient had the iCare HOME and the corresponding times at which the measurements were taken three months prior to the glaucoma stent procedure OD. Notably, the highest IOP measurements were recorded overnight and in the morning, whereas the lowest IOP measurements were recorded in the afternoon and early evening.
The decision was made to perform additional surgery in the right eye three months after those initial iCare measurements were taken to better control IOP fluctuations, as the patient had pericentral visual field loss OD (Figure 2) and diffuse optic nerve thinning temporally OD (Figure 3).
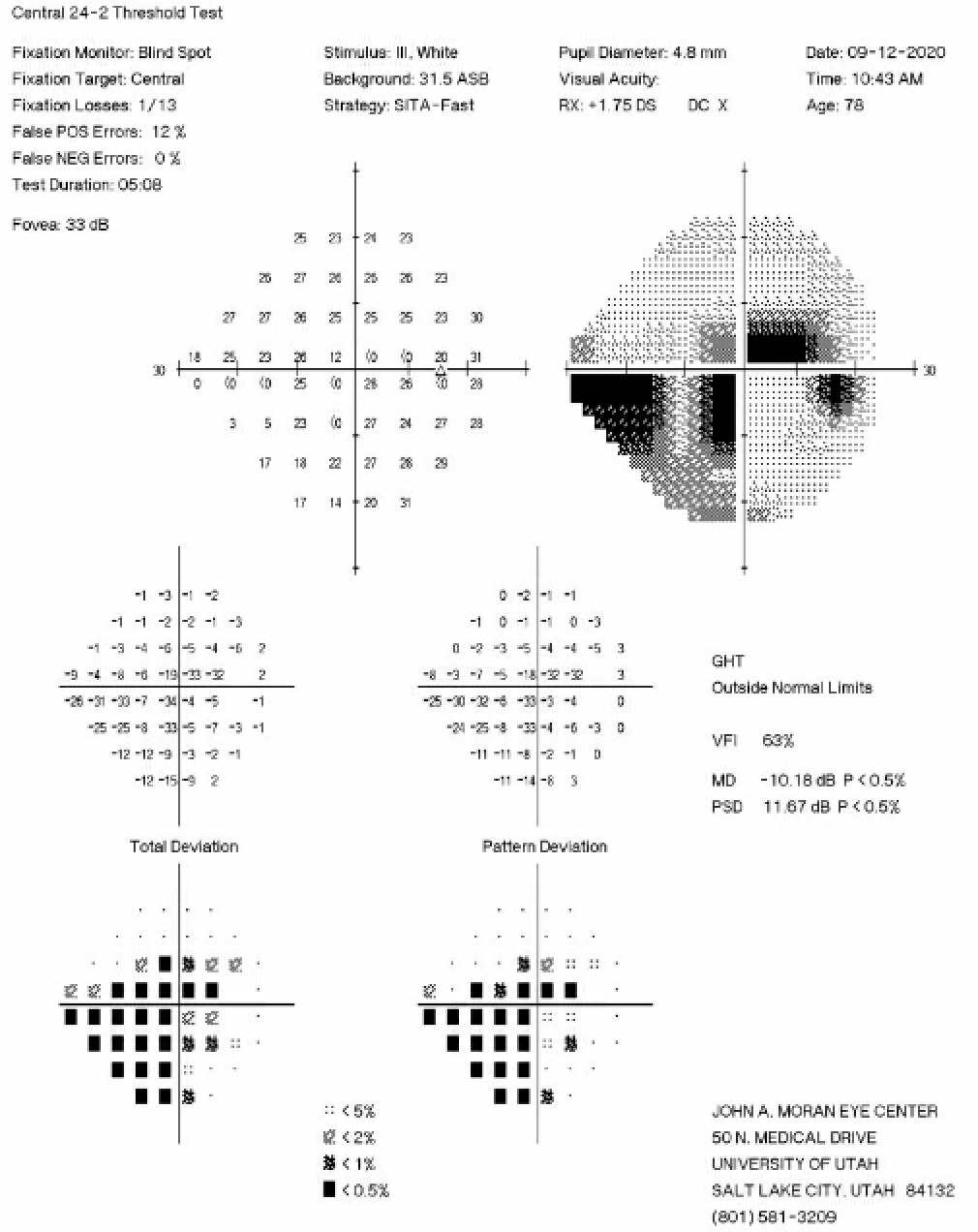
Figure 2: 24-2 Humphrey Visual Field test from six months prior to the initial iCare HOME measurements showing an inferior nasal step and pericentral visual field loss in the right eye.
Nine months after the XEN gel stent with mitomycin C/Ologen was placed in the right eye, the patient was asked to repeat the iCare measurements. A summary of the results is shown in Figure 4 below.

Figure 4: Summary of the results of iCare HOME measurements taken nine months after the glaucoma stent procedure OD. All the measurements were “excellent” in quality. Average IOP and variability of IOP in the right eye were now both lower compared to the iCare HOME measurements from one year prior.
Discussion:
IOP varies both daily and hourly, and clinicians are not able to fully understand patients’ IOP when only capturing measurements at office visits. In a study conducted at the Wilmer Eye Institute, it was found that mean IOP was slightly lower by home tonometry readings than by clinic readings alone, while IOP fluctuation and IOP spikes were significantly higher than those measured in clinic.4 Additionally, those researchers found that mean daily measurements exceeded the recent clinic maximum IOP in 44% of patients, and that mean daily measurements by home iCare were greater than any historic IOP measured in clinic in 13% of patients.4 Furthermore, the peak home IOP occurred outside of the typical 8:00 am – 5:00 pm office hours on half of the days.4 Diurnal IOP fluctuations have been documented dating back several decades. In one study analyzing 2272 diurnal curves of IOP measurements, 41% of the peaks were found at the early morning IOP measurement, while 24% of peaks were found at the second, mid-morning measurement.5 Another study showed this pattern of nocturnal IOP elevation occurs independently of central corneal thickness, corneal hysteresis, and corneal resistance factor.6
Mean IOP and diurnal fluctuation in IOP are typically higher in XFG than in POAG, and reduction of these measures has been documented to be more effective in XFG than in POAG in preventing visual field damage.7 In our patient, implanting a XEN gel stent with mitomycin C/Ologen in the eye with significant glaucoma damage lowered her average home iCare reading in that eye from 18.2 to 9.5, and the standard deviation of IOP measures decreased from 3.6 to 1.0. With these significantly improved eye pressures, RNFL thickness and visual fields have remained relatively stable for the past two years since her surgery. This case demonstrates the importance of understanding how IOP varies throughout the day, and the efficacy of surgical intervention on minimizing both fluctuations in these measurements and glaucomatous changes in the eye.
References:
-
- Ritch R, Schlötzer-Schrehardt U. Exfoliation syndrome. Surv Ophthalmol. 2001;45(4):265-315. doi:10.1016/s0039-6257(00)00196-x
- Yüksel N, Yılmaz Tuğan B. Pseudoexfoliation Glaucoma: Clinical Presentation and Therapeutic Options. Turk J Ophthalmol. 2023;53(4):247-256. doi:10.4274/tjo.galenos.2023.76300
- Pompoco CJ, Curtin K, Taylor S, et al. Summary of Utah Project on Exfoliation Syndrome (UPEXS): using a large database to identify systemic comorbidities. BMJ Open Ophthalmol. 2021;6(1):e000803. doi:10.1136/bmjophth-2021-000803
- McGlumphy EJ, Mihailovic A, Ramulu PY, Johnson TV. Home Self-tonometry Trials Compared with Clinic Tonometry in Patients with Glaucoma. Ophthalmol Glaucoma. 2021;4(6):569-580. doi:10.1016/j.ogla.2021.03.017
- David R, Zangwill L, Briscoe D, Dagan M, Yagev R, Yassur Y. Diurnal intraocular pressure variations: an analysis of 690 diurnal curves. Br J Ophthalmol. 1992;76(5):280-283.
- Bagga H, Liu JHK, Weinreb RN. Intraocular pressure measurements throughout the 24 h. Curr Opin Ophthalmol. 2009;20(2):79-83. doi:10.1097/ICU.0b013e32831eef4f
- Vahedian Z, Salmanroghani R, Fakhraie G, et al. Pseudoexfoliation syndrome: Effect of phacoemulsification on intraocular pressure and its diurnal variation. J Curr Ophthalmol. 2015;27(1-2):12-15. doi:10.1016/j.joco.2015.09.006
Identifier: Moran_CORE_127006
Copyright: Tara Gallant and Barbara Wirostko ©2023. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Neuropathic Ocular Pain
Home / Neuro-Ophthalmology / Headaches and Positive Visual Phenomena
Title: Neuropathic Ocular Pain
Authors: Jaxon J. Huang, 4th year medical student, University of Hawaii; Anat Galor, MD MSPH, University of Miami, Bascom Palmer Eye Institute
Date: 8/15/2023
Keywords/Main Subjects: neuropathic ocular pain, eye pain, headache, photophobia, dry eye
Diagnosis: Neuropathic ocular pain
Description of Case:
A 49-year-old female with a history of depression, fibromyalgia, and traumatic brain injury presented to the ophthalmology clinic for dry eye symptoms of irritation and grittiness in both eyes for 2 years. She also reported burning and aching eye pain, frequent headaches, and sensitivity to light and wind. The patient had been using artificial tears 5 times a day for the past 6 months with no relief in symptoms. Her dry eye and ocular pain symptoms were evaluated using questionnaires, including the 5 Item Dry Eye Questionnaire (DEQ5; scale 0-22)1, Ocular Surface Disease Index (OSDI; scale 0-100)2, and the Neuropathic Pain Symptom Inventory modified for the Eye (NPSI-Eye; scale 0-40)3. Her baseline scores were as follows: DEQ5=16 (severe), OSDI=87.5 (severe), NPSI-Eye=27 (severe).
Examination:
- InflammaDry tear test (Quidel, San Diego)4: negative OD; negative OS
- Upper eyelid laxity determined by rotation (0=0-25%; 1=25-50%; 2=50-100%)5: 0 OD; 0 OS
- Lower eyelid laxity determined by snap back test (0=prompt snapback; 1=slowed return; 2=does not return until blinking)5: 0 OD; 0 OS
- Anterior blepharitis (0=none; 1=mild; 2=moderate; 3=severe)6: 1 OD; 1 OS
- Telangiectasias of the lower eyelids (0=none; 1=mild; 2=moderate; 3=severe)6: 0 OD; 1 OS
- Inferior meibomian gland plugging (0=none; 1=less than 1/3; 2=between 1/3 and 2/3; 3=greater than 2/3 lid involvement)6: 1 OD; 1 OS
- Tear break-up time (TBUT)7: 8 OD; 9 OS
- Conjunctivochalasis assessed nasally, medially, and temporally (0=none; 1=mild; 2=moderate; 3=severe)7: 1/0/1 OD; 1/0/1 OS
- Corneal staining assessed inferiorly, nasally, superiorly, temporally, and centrally then summed (0=none; 1=mild; 2=moderate; 3=severe)7: 4 OD; 3 OS
- Ocular pain before anesthesia (scale of 0-10; 0=no pain; 10=most severe pain)8: 7
- Ocular pain after anesthesia (scale of 0-10; 0=no pain; 10=most severe pain)8: 8
- Schirmer’s test (mm of wetting at 5 minutes)7: 20 OD; 9 OS
- Meibum quality (0=clear; 1=cloudy; 2=granular; 3=toothpaste; 4=no meibum extracted)6: 0 OD; 0 OS
- Inferior eyelid meibomian gland dropout graded to the Meiboscale (0=no dropout; 1=<25% dropout; 3=25% to 75% dropout; 3=>75% dropout)9: 1 OD; 1 OS
Imaging:
- In-vivo confocal microscopy10: 5+ activated dendritic cells, increased corneal nerve tortuosity, decreased corneal nerve density
Images:

Figure 1. In-vivo confocal microscopy image of corneal nerves showing activated dendritic cells (white dashed circles) and increased nerve tortuosity with decreased nerve density (white arrow).
Clinical Course:
The patient was diagnosed with neuropathic ocular pain. She was treated with 35 units of Botulinum toxin A (BoNT-A) based on a modified migraine protocol targeting the corrugator, procerus, and the frontalis muscles.11 To target photophobia, the patient was prescribed FL-41 tinted lenses. While she still reported occasional symptoms of ocular irritation, the frequency and severity of her ocular pain and migraines were reduced. Additionally, she endorsed a reduction in photophobia with use of the FL-41 tinted lenses. Her post-treatment questionnaire scores evaluating dry eye and ocular pain symptoms were as follows: DEQ5=10 (mild-moderate), OSDI=79 (severe), NPSI-Eye=17 (moderate).
Discussion:
Ocular pain is frequently incorporated under the term “dry eye disease (DED)”, which encompasses a variety of symptoms such as dryness, irritation, and pain, along with clinical signs of decreased tear production or loss of tear film homeostasis. DED pain symptoms in particular can be driven by nociceptive sources, such as ocular surface inflammation and tear film instability, or by neuropathic sources, such as nerve dysfunction.10, 12, 13 Currently, a diagnosis of a neuropathic contributor to ocular pain is made based on clinical examination findings. Individuals with neuropathic ocular pain (NOP) often describe their pain as “burning” or “shooting” and endorse evoked pain to triggers such as wind and light.14 Additionally, individuals will often present with a discordance between pain symptoms and clinical signs of tear dysfunction, with symptoms that are out of proportion to signs.15 Furthermore, persistent pain after application of a topical anesthetic and abnormal corneal nerve anatomy and function may be observed in NOP.8,16 Individuals with NOP often fail treatments targeted towards improving tear health (e.g. artificial tears and anti-inflammatory drops) and have co-morbid pain conditions, including migraine and fibromyalgia.17, 18
The current treatment of NOP involves targeting underlying nerve dysfunction using topical, oral, or adjuvant therapy. Topical therapies include autologous serum tears and transient receptor potential vanilloid (TRPV1) antagonists19, while oral agents include gabapentin, pregabalin, and nortriptyline, all of which have varying success in treating NOP.20,21 Adjuvant therapies consist of transcutaneous electrical nerve stimulation devices and BoNT-A injections, which have both been shown to decrease symptoms of ocular pain and photophobia.22, 23 In addition, the use of FL-41 tinted lenses has been reported to be helpful in alleviating photophobia and migraine.24 However, despite the wide variety of treatment options available, pain persists in many NOP patients, highlighting the need for new therapeutic approaches.25
Summary of the Case:
NOP is diagnosed clinically based on findings of “burning” pain, pain evoked by wind or light, symptoms that outweigh clinical signs of tear dysfunction, persistent pain after topical anesthetic, and abnormal corneal anatomy and function. While various topical, oral, and adjuvant therapies have been investigated and shown to have varying efficacy, further studies are needed to develop novel approaches for patients who do not respond to current options.
References:
- Chalmers RL, Begley CG, Caffery B. Validation of the 5-Item Dry Eye Questionnaire (DEQ-5): Discrimination across self-assessed severity and aqueous tear deficient dry eye diagnoses. Cont Lens Anterior Eye 2010;33(2):55-60.
- Schiffman RM, Christianson MD, Jacobsen G, et al. Reliability and validity of the Ocular Surface Disease Index. Arch Ophthalmol 2000;118(5):615-21.
- Farhangi M, Feuer W, Galor A, et al. Modification of the Neuropathic Pain Symptom Inventory for use in eye pain (NPSI-Eye). Pain 2019;160(7):1541-50.
- Sambursky R, Davitt WF, 3rd, Latkany R, et al. Sensitivity and specificity of a point-of-care matrix metalloproteinase 9 immunoassay for diagnosing inflammation related to dry eye. JAMA Ophthalmol 2013;131(1):24-8.
- Ansari Z, Singh R, Alabiad C, Galor A. Prevalence, risk factors, and morbidity of eye lid laxity in a veteran population. Cornea 2015;34(1):32-6.
- Foulks GN, Bron AJ. Meibomian gland dysfunction: a clinical scheme for description, diagnosis, classification, and grading. Ocul Surf 2003;1(3):107-26.
- Methodologies to diagnose and monitor dry eye disease: report of the Diagnostic Methodology Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf 2007;5(2):108-52.
- Crane AM, Feuer W, Felix ER, et al. Evidence of central sensitisation in those with dry eye symptoms and neuropathic-like ocular pain complaints: incomplete response to topical anaesthesia and generalised heightened sensitivity to evoked pain. Br J Ophthalmol 2017;101(9):1238-43.
- Pult H, Riede-Pult B. Comparison of subjective grading and objective assessment in meibography. Cont Lens Anterior Eye 2013;36(1):22-7.
- Belmonte C, Nichols JJ, Cox SM, et al. TFOS DEWS II pain and sensation report. Ocul Surf 2017;15(3):404-37.
- Venkateswaran N, Hwang J, Rong AJ, et al. Periorbital botulinum toxin A improves photophobia and sensations of dryness in patients without migraine: Case series of four patients. Am J Ophthalmol Case Rep 2020;19:100809.
- Bron AJ, de Paiva CS, Chauhan SK, et al. TFOS DEWS II pathophysiology report. Ocul Surf 2017;15(3):438-510.
- Craig JP, Nichols KK, Akpek EK, et al. TFOS DEWS II Definition and Classification Report. Ocul Surf 2017;15(3):276-83.
- Kalangara JP, Galor A, Levitt RC, et al. Characteristics of Ocular Pain Complaints in Patients With Idiopathic Dry Eye Symptoms. Eye Contact Lens 2017;43(3):192-8.
- Ong ES, Felix ER, Levitt RC, et al. Epidemiology of discordance between symptoms and signs of dry eye. Br J Ophthalmol 2018;102(5):674-9.
- Galor A, Felix ER, Feuer W, et al. Corneal Nerve Pathway Function in Individuals with Dry Eye Symptoms. Ophthalmology 2021;128(4):619-21.
- Galor A, Batawi H, Felix ER, et al. Incomplete response to artificial tears is associated with features of neuropathic ocular pain. Br J Ophthalmol 2016;100(6):745-9.
- Lee Y, Kim M, Galor A. Beyond dry eye: how co-morbidities influence disease phenotype in dry eye disease. Clin Exp Optom 2022;105(2):177-85.
- Patel S, Mittal R, Sarantopoulos KD, Galor A. Neuropathic ocular surface pain: Emerging drug targets and therapeutic implications. Expert Opin Ther Targets 2022;26(8):681-95.
- Small LR, Galor A, Felix ER, et al. Oral Gabapentinoids and Nerve Blocks for the Treatment of Chronic Ocular Pain. Eye Contact Lens 2020;46(3):174-81.
- Ozmen MC, Dieckmann G, Cox SM, et al. Efficacy and tolerability of nortriptyline in the management of neuropathic corneal pain. Ocul Surf 2020;18(4):814-20.
- Sivanesan E, Levitt RC, Sarantopoulos CD, et al. Noninvasive Electrical Stimulation for the Treatment of Chronic Ocular Pain and Photophobia. Neuromodulation 2018;21(8):727-34.
- Reyes N, Huang JJ, Choudhury A, et al. Botulinum toxin A decreases neural activity in pain-related brain regions in individuals with chronic ocular pain and photophobia. Front Neurosci 2023;17:1202341.
- Katz BJ, Digre KB. Diagnosis, pathophysiology, and treatment of photophobia. Surv Ophthalmol 2016;61(4):466-77.
- Siedlecki AN, Smith SD, Siedlecki AR, et al. Ocular pain response to treatment in dry eye patients. Ocul Surf 2020;18(2):305-11.
Faculty Approval by: Griffin Jardine, MD
Identifier: Moran_CORE_126997
Copyright: Jaxon J. Huang and Anat Galor ©2023. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Case Report and Clinical Features of Elschnig Pearls
Home / Lens and Cataract / Complications of Cataract Surgery
Title: Case Report and Clinical Features of Elschnig Pearls
Authors: Marissa Larochelle, MD; Wyatt Corbin, BS
Date: 9/22/2023
Keywords/Main Subjects: Elschnig pearl, cataract surgery, PCO, posterior capsular opacification
Diagnosis: Elschnig Pearls
Images or video:

Image 1. Slit-lamp exam of the patient’s left eye’s anterior surface. Cloudy material consisting of Elschnig pearls is protruding from the superonasal and inferotemporal quadrants and obscuring the visual axis. Some of the material may also be seen extruding anteriorly from behind the inferotemporal iris at the 5 o’clock position. The iris is poorly dilated and moderate iris bombe is also present.

Image 2. Magnified image of a large amount of round, individually translucent yet collectively opaque Elschnig pearls protruding from the superonasal and inferotemporal quadrants and obscuring the visual axis; PCIOL is present and appears centered. Individual Elschnig pearls may be visualized in this image.

Image 3. Slit-lamp exam of the patient’s left eye’s anterior surface after surgical peeling and aspiration of the Elschnig pearls. Unlike the comparative preoperative photo, no Elschnig pearls are seen in this image and the visual axis appears clear.
Case Report
Patient medical history:
A 67-year-old female was referred for the surgical treatment of Elschnig pearls. A uveitis colleague noted decreased visual acuity due to Elschnig pearls on slit lamp exam one month prior. The patient described having “filmy vision” in her left eye with light sensitivity. The visual acuity in the left eye was 20/40 with no improvement with pinhole testing, a decrease from her baseline vision of 20/20 documented 3 months prior. Ultrasound biomicroscopy confirmed the presence of material on the anterior surface of the IOL optic consistent with our clinical diagnosis.
The patient has an ocular history of pseudophakia of both eyes, non-granulomatous anterior uveitis in both eyes, and intermediate uveitis in her left eye. Cataract surgery was performed nearly 17 years prior in the left (affected eye). For her ocular inflammatory conditions, she was taking Prednisolone BID OU, methotrexate 25mg weekly, and folic acid 1mg daily. The patient also has a history of type 2 diabetes mellitus, hypothyroidism, hypertension, chronic sinusitis, cervicalgia, and headache.
Examination:
The patient presented with a visual acuity of 20/20 in the right eye and a visual acuity of 20/40 with no improvement with pinhole testing in the left eye. At the current visit, she had a normal eye pressure reading of 10 in the left eye. The patient did not have any afferent pupillary defects, although the left eye had minimal reactivity to light. All extraocular movements were intact.
The left eye presented with extensive inferotemporal and superonasal Elschnig pearls anterior and posterior to the PCIOL and obscuring the visual axis (see Images 1 and 2). The Elschnig pearls were also seen extruding anteriorly from behind the inferotemporal iris at the 5 o’clock position (see Image 1). The left eye also had old pigmented keratic precipitates inferiorly, a quiet anterior chamber and a centered posterior chamber intraocular lens. Otherwise, the external and fundus exams were normal.
Management:
Due to the extensive amount of Elschnig pearls in the visual axis, the patient was managed with anterior chamber washout via surgical peeling and aspiration (see Image 3). This was done in the operating room with bimanual irrigation and aspiration. She was treated prophylactically with Prednisolone acetate 1% drops four times daily starting 1 week before the surgery to prevent a uveitis flare in the peri-operative period.
After a successful surgery, the patient’s visual axis became clear. However, her visual acuity did not improve. We suspect that the patient’s lack of visual improvement post-operatively may be due to their long-standing history of uveitis.
Clinical Features of Elschnig Pearls
Definition and Background Information: Elschnig pearls are cystic, giant cell-like structures that are thought to be a manifestation of regenerative posterior capsular opacification (PCO) caused by residual equatorial lens epithelial cell (LEC) migration and proliferation between the posterior capsule and the intraocular lens (IOL) after cataract surgery.1 The exact morphology and histology of Elschnig pearls are yet to be fully elucidated, but they have been described as containing a nucleus and few cell organelles.1,2 Elschnig pearls are also thought to be products of the ballooning of cytoplasm emerging from the cell membrane of degenerating lens fibers.1,3 Interestingly, another case report of Elschnig pearls in a patient with chronic uveitis was published, suggesting a possible mechanism of development associated with intraocular inflammation.4 However, one study reported that individual variability may have a greater effect on their formation than the degree of inflammation.4,5
The epidemiology of Elschnig pearls is unknown because they have a variable presentation and thus are difficult to distinguish from other forms of PCO.4 For example, our patient presented with debris, which was revealed to be Elschnig pearls, posterior and anterior to the IOL optic, rather than the more common clinical presentation of PCO posterior to IOL optics. Also, although they were once considered rare, they are now regarded as a relatively common post-operation complication of cataract surgery.4,6 It is not known exactly why they are regarded as a more common complication now. However, it may be due to enhanced diagnostic techniques including ultrasound biomicroscopy increasing their detection rate, or another factor related to the current population or modern cataract surgery practices resulting in increased Elschnig pearl formation. More research would be needed to elucidate why their prevalence has seemed to increase.
Diagnosis: Like other forms of PCO, Elschnig pearls are diagnosed in clinical settings via slit-lamp microscopy. They appear as cloudy clusters of pearls, most commonly posterior to PCIOLs and anterior to the posterior lens capsule. However, they may also present anterior to PCIOLs yet mostly contained within the posterior chamber such as in our patient.
Symptoms: Like other forms of PCO, Elschnig pearls do not always cause symptoms, but they can lead to a decrease in visual acuity and contrast sensitivity due to light scattering. It is proposed that the cellular material inside the Elschnig pearls has a higher refractive index which results in this light scattering.1,4,7-9
Prognosis: The presence of Elschnig pearls generally does not resolve spontaneously, although there are case reports of rare spontaneous regression.10,11 In part, spontaneous regression is rare because when visual symptoms occur the pearls are often treated immediately by neodymium yttrium garnet (Nd:YAG) laser capsulotomy. Like other forms of PCO, however, Elschnig pearls may continually progress or remain stable over time.1,4,12 Regression, with or without the assistance of laser or surgical intervention, generally occurs through pearls falling through a capsulotomy posteriorly into the vitreous, phagocytosis of the pearls by macrophages, or apoptotic cell death.6,12 Although the presence of Elschnig pearls in a patient is generally persistent for months to years, one study demonstrated that individual pearls may appear and disappear within days, most likely via different mechanisms as stated earlier.1 The progression and regression of individual pearls were observed to be influenced by the size, shape, and solidity of the pearls, as well as variability between patients.1,4,5
Management: Similar to other forms of PCO, Elschnig pearls may only need to be treated if visual symptoms occur, thus patient monitoring with patient reports, functional vision testing, and slit-lamp examination is a part of management. For example, although one case report presented a patient with Elschnig pearls obstructing his central visual axis, his vision was still 20/15, so no treatment was performed.10 When symptomatic, the primary treatment option for the pearl or regeneratory form of PCO is Nd:YAG laser capsulotomy due to the convenience of the procedure and decreased risks associated with surgical treatment. Additionally, an anterior chamber washout by surgical peeling, irrigation, and aspiration may be performed.13 Notably, the fibrous form of PCO is only treated by Nd:YAG laser capsulotomy.13 Patients with other complications secondary or unrelated to the Elschnig pearls, prior cataract surgery, or potential underlying causes of inflammation, such as uveitides causing synechiae, must also be managed via the appropriate surgical or medical interventions. Chronic medically induced pupil dilation to allow for a greater visual window and bimanual anterior vitrectomy may also be considered as treatment options for patients with Elschnig pearls.
One study compared Nd:YAG laser capsulotomy with surgical peeling and aspiration and showed that both techniques are comparable with regard to visual outcomes.13 However, Nd:YAG laser capsulotomy was associated with a higher incidence of spikes in intraocular pressure (IOP) and retinal detachment whereas treatment with surgical peeling and aspiration was associated with a higher incidence of pearl recurrence. Thus, caution must be exercised when considering Nd:YAG laser capsulotomy in patients with previous retinal disease and pathologic myopia.13
After Nd:YAG laser capsulotomy or surgical peeling, irrigation, and aspiration, the patient must be monitored to diagnose and treat any post-operative inflammation or spikes in IOP. Repeat treatment may also be necessary to treat recurring Elschnig pearls by utilizing any of the aforementioned methods based on clinical judgment.
Summary of the Case:
We present a case and two images of Elschnig pearls in a 67-year-old female patient with a history of pseudophakia in her ipsilateral eye and anterior segment uveitis. Given this patient’s clinical background and the extent of growth of Elschnig pearls, an anterior chamber washout with surgical peeling, irrigation, and aspiration was performed successfully.
Elschnig pearls are a relatively common regenerative form of PCO after cataract surgery and are currently thought to be cystic, cell-like structures that are products of lens fiber cells that degenerate after cataract surgery. The most common risk factor for Elschnig pearls is previous cataract surgery, although chronic inflammation and other patient-specific factors may present varying risks for pearl formation. Patients with Elschnig pearls may experience visual disturbances such as photopsias which are commonly persistent. They are primarily treated with Nd:YAG laser capsulotomy. Although if clinically appropriate, they may also be treated surgically with an anterior chamber washout via irrigation and aspiration. Each treatment modality is accompanied by risks and benefits that must be weighed by each clinician when considering the safest and most optimal treatment for specific patients.
Format: Case Report and Clinical Features
References:
-
- Findl O, Neumayer T, Hirnschall N, Buehl W. Natural Course of Elschnig Pearl Formation and Disappearance. Investigative Opthalmology & Visual Science. 2010;51(3):1547. doi:10.1167/iovs.09-3989
- COWAN A, FRY. SECONDARY CATARACT. Archives of Ophthalmology. 1937;18(1):12. doi:10.1001/archopht.1937.00850070024002
- Jongebloed WL, Kalicharan D, Los LI, van der Veen G, Worst JG. A combined scanning and transmission electronmicroscopic investigation of human (secondary) cataract material. Doc Ophthalmol. 1991;78(3-4):325-334. doi:10.1007/BF00165696
- K Foutch B, A Garcia C, S Ferguson A. Pearls of Elschnig. J Ophthalmic Vis Res. 2019;14(4):525-527. doi:10.18502/jovr.v14i4.5469
- Neumayer T, Buehl W, Findl O. Effect of topical prednisolone and diclofenac on the short-term change in morphology of posterior capsular opacification. Am J Ophthalmol. 2006;142(4):550-556. doi:10.1016/j.ajo.2006.04.047
- Caballero A, Salinas M, Marin JM. Spontaneous disappearance of Elschnig pearls after neodymium:YAG laser posterior capsulotomy. J Cataract Refract Surg. 1997;23(10):1590-1594. doi:10.1016/s0886-3350(97)80035-1
- Brown N. Visibility of transparent objects in the eye by retroillumination. Br J Ophthalmol. 1971;55(8):517-524. doi:10.1136/bjo.55.8.517
- Buehl W, Sacu S, Findl O. Association between intensity of posterior capsule opacification and contrast sensitivity. Am J Ophthalmol. 2005;140(5):927-930. doi:10.1016/j.ajo.2005.05.022
- Jose RMJ, Bender LE, Boyce JF, Heatley C. Correlation between the measurement of posterior capsule opacification severity and visual function testing. J Cataract Refract Surg. 2005;31(3):534-542. doi:10.1016/j.jcrs.2004.07.022
- Nakashima Y, Yoshitomi F, Oshika T. Regression of Elschnig pearls on the posterior capsule in a pseudophakic eye. Arch Ophthalmol. 2002;120(3):397-398.
- Caballero A, Marín JM, Salinas M. Spontaneous regression of Elschnig pearl posterior capsule opacification. J Cataract Refract Surg. 2000;26(5):779-780. doi:10.1016/s0886-3350(00)00412-0
- Kurosaka D, Kato K, Kurosaka H, Yoshino M, Nakamura K, Negishi K. Elschnig pearl formation along the neodymium:YAG laser posterior capsulotomy margin. Long-term follow-up. J Cataract Refract Surg. 2002;28(10):1809-1813. doi:10.1016/s0886-3350(02)01222-1
- Bhargava R, Kumar P, Sharma SK, Kaur A. A randomized controlled trial of peeling and aspiration of Elschnig pearls and neodymium: yttrium-aluminium-garnet laser capsulotomy. Int J Ophthalmol. 2015;8(3):590-596. doi:10.3980/j.issn.2222-3959.2015.03.28
Faculty Approval by: Austin Nakatsuka, MD
Copyright statement: Marissa Larochelle and Wyatt Corbin ©2023. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
A Brief Overview of Salzmann’s Nodular Degeneration and Its Topographical Outcomes
Home / External Disease and Cornea / Clinical Approach to Depositions and Degenerations of the Conjunctiva, Cornea, and Sclera
Author: Tanmay Majmudar, BS, MS4 at Drexel University College of Medicine
Photographer: James Gilman, CRA FOPS
Date: 9/12/2023
Keywords/Main Subject: cornea, corneal dystrophy, corneal topography, Salmann’s nodular degeneration, superficial keratectomy
Diagnosis: Salzmann’s Nodular Degeneration
Case Presentation:
A 64-year-old female was referred to the cornea clinic by an outside optometrist for progressively worsening vision in both eyes (OS worse than OD). She had a remote history of pterygium removal in both eyes, high astigmatism and hyperopia corrected with bifocal prescription glasses, and early dry stage nonexudative age-related macular degeneration. She also used preservative free artificial tears twice daily for chronic eye dryness and bedtime lubricant ointment as needed. Her past medical, family, and social histories were otherwise unremarkable.
Upon presentation to our clinic, her best corrected visual acuity (BCVA) with prescription bifocals was 20/40-1 and 20/60 in the right and left eyes respectively. Slit lamp examination of the anterior segment revealed a 2.5 mm greyish nodule (see a representative slit-lamp image from another patient with the same diagnosis in Figure 1) between the corneal epithelium and Bowman’s layer nasally over the pterygium scar in the right eye and a similar appearing 4 mm nodule nasally in the left eye as well. Trace to 1+ superficial punctate keratitis stable from prior exams was also noted in both eyes. The rest of the exam, including a dilated fundus exam, was unremarkable besides nuclear sclerotic cataracts (graded 2+) in both eyes.

Figure 1. Slit-lamp image of another patient with Salzmann’s nodular degeneration showing the characteristic blue-greyish nodules. Image taken by James Gilman, University of Utah John A Moran Eye Center.
Corneal Topography:
Atlas corneal topography was ordered to characterize the corneal nodules seen on slit-lamp examination, especially given the clinical history of astigmatism. Figure 2 shows a Zeiss Atlas report of the patient for both eyes with an axial curvature map and Placido disc-based “rings image”. Topography in the left eye shows a high irregular astigmatism with focal steepening and irregular mires noted in the superotemporal region. A crab-claw like pattern of irregular astigmatism is more noticeable nasally in the right eye. These focal areas of astigmatism largely correspond to the location of the blue-grayish nodules noted on slit-lamp examination.

Figure 2. Pre-operative corneal topography.
Diagnosis:
A diagnosis of Salzmann’s nodular degeneration (SND) was made on the basis of the characteristic appearance on slit-lamp examination, clinical history of bilateral, gradually progressive, painless visual decline, history of prior ocular surface surgery (pterygium removal) and chronic dry eyes, and high irregular astigmatism noted on corneal topography corresponding to areas of nodular growth.
Treatment & Clinical Course:
To improve visual acuity and to reduce the high irregular astigmatism, a superficial keratectomy (SK) was recommended to remove the Salzmann’s nodules; however, the patient was informed that refractive outcomes may not eliminate her astigmatism and that recurrence of these nodules was possible.
Post-Operative Outcomes:
About two weeks after her initial presentation to the cornea clinic, the patient underwent SK on her left eye. At the time of writing, post-operative refractive and topographical data for the patient’s right eye (surgery completed about two weeks after the left eye) are unavailable. Two months postoperatively, her left cornea’ epithelium healed with complete clearing of the subepithelial opacities. Post-operatively her left eye achieved a best corrected visual acuity of 20/20 -2, with substantial reduction in astigmatic error (Figure 3, Table 1).
Table 1. Pre- and post-operative refraction in a patient with Salzmann’s nodular degeneration.
| Pre-operative Manifest Refraction (OS) | |
| -0.25 + 1.50 x 045 | VA 20/60 |
| Post-operative Manifest Refraction | |
| -0.50 + 1.00 x 030 | VA 20/20 -2 |

Figure 3. Left eye topography before (top row) and after (bottom row) superficial keratectomy for removal of Salzmann’s nodules. After intervention, topography shows a more spherical cornea with substantially more regular mires and corresponding reduction in both regular and irregular astigmatism from 5.36 diopters to 1.00 D.
Discussion:
Salzmann’s Nodular Degeneration (SND) is an unusual and relatively rare1 disorder of the cornea that involves the growth of blue-grayish fibrous corneal opacities of various sizes (typically 1-3 mm) anterior to the Bowman’s capsule2. It is a slowly progressive, noninflammatory degenerative condition that typically affects middle-aged Caucasian females (between ages 50-60)2,3 and occurs bilaterally3. Though many patients with SND are asymptomatic early in their disease course, gradually decreasing visual acuity due to irregular astigmatism, hyperopic shifts, or mechanical tear film disturbances is the most common presenting symptom3-5. Other symptoms are related ocular surface irritation, including foreign body sensation, pain, tearing, blepharospasms, or recurrent corneal erosions are also reported2,3,6.
The precise mechanism of this degeneration is poorly understood. Mechanical disruption between the corneal epithelium and stroma from chronic ocular surface inflammatory conditions such as dry eye, meibomian gland dysfunction (MGD), long-term contact lens wear, keratoconjunctivitis sicca, exposure keratitis, pterygium amongst others have been implicated in prior studies2,3,6-9. SND has also been observed in the setting of previous ocular surgery and trauma, such as after LASIK5,10, cataract extraction11, radial keratotomy12, and corneal transplants13. Our patient’s history of chronic eye disease, pterygium removal, and long-term contact lens wear also supports the pathogenic role of these risk factors in the development of SND. Finally, prior studies have documented associations between systemic inflammatory conditions, such as Crohn’s disease14, and SND while others have also attempted to elucidate a genetic involvement15.
Diagnosis of SND is usually made based on a careful examination of corneal structure under slit-lamp biomicroscopy. Nodules appear as blueish-white to grayish-yellow subepithelial elevations that may occasionally stain with fluorescein2. While diagnosis of SND is primarily based on slit-lamp examination findings and the clinical history, it may be supported by corneal topography, as in this patient, or high-frequency ultrasound biomicroscopy, in-vivo confocal microscopy, and optical coherence tomography of the anterior segment. Classic histologic features include a thinned epithelium overlying the nodules, fragmented or absent Bowman’s membrane, or stromal scarring2,16. Unfortunately, these histologic features of non-specific and can frequently be seen with old corneal scars from trauma or inflammation.
Management options are largely driven by the severity of symptoms. Although asymptomatic patients are primarily observed, treatment of mild disease is aimed at addressing the underlying etiology through medical management. Use of preservative-free lubricant eye drops, lid hygiene, and warm compresses in patients with chronic dry eye has been effective in the majority of cases3. Medical options to treat SND associated with chronic ocular inflammation include steroids, NSAIDs, cyclosporine and doxycycline amongst others2. If nodules occur in the setting of contact lens use, then reduction of use or cessation may also relieve symptoms. However, in cases that do not respond to conservative management or those patients with visually significant nodules, surgical management is necessary.
A variety of surgical options exist, including SK, otherwise known as Salzmann nodulectomy, phototherapeutic keratectomy (PTK), lamellar or penetrating keratoplasty. Of these, SK is the most common method for nodule removal. Briefly, the epithelium overlying the nodules is removed and the nodular edge is simply peeled off the corneal surface with forceps or a flat blade, leaving Bowman’s layer almost untouched2. Post-operative outcomes are very favorable in reducing astigmatic error and improvements in visual acuity have been recorded in 79.2%-100% of patients in prior studies2,3,6. In rare cases, persistent anterior stromal haze may necessitate further treatment with PTK. Rarely, lamellar or penetrating keratoplasty may be required for patients with concurrent corneal pathologies involving the deeper stroma and endothelium. Although varying rates of recurrences have been reported3,6,7, visually significant recurrences are uncommon (ranging from 5 to 20%) and can be further minimized with management of the underlying etiologies3,6.
Beyond the visual limitations of corneal nodularity itself, the accuracy of keratometry and biometry calculations, a critical step in selecting the appropriate intraocular lens (IOL), is largely based on a clear cornea with a smooth regular surface. Patients with corneal dystrophies, such as SND or epithelial basement membrane dystrophy, have irregular corneas that can such adversely affect biometry, recommended IOL choice, and postoperative visual outcomes. In patients requiring cataract surgery who also have comorbid corneal irregularities, prior studies have showed improved reliability in biometry measurements after management with SK and better post-cataract optical quality17,18. Although our patient, with 2+ nuclear sclerotic cataracts, continues to deny visually limiting symptoms related to them, this case nonetheless highlights the importance of addressing the refractive sequelae of SND in consideration for future cataract surgery.
References:
- Gupta PK, Drinkwater OJ, VanDusen KW, Brissette AR, Starr CE. Prevalence of ocular surface dysfunction in patients presenting for cataract surgery evaluation. J Cataract Refract Surg. Sep 2018;44(9):1090-1096.
- Hamada S, Darrad K, McDonnell PJ. Salzmann’s nodular corneal degeneration (SNCD): clinical findings, risk factors, prognosis and the role of previous contact lens wear. Cont Lens Anterior Eye. Aug 2011;34(4):173-8.
- Graue-Hernández EO, Mannis MJ, Eliasieh K, et al. Salzmann nodular degeneration. Cornea. Mar 2010;29(3):283-9.
- Oster JG, Steinert RF, Hogan RN. Reduction of hyperopia associated with manual excision of Salzmann’s nodular degeneration. J Refract Surg. Jul-Aug 2001;17(4):466-9.
- VanderBeek BL, Silverman RH, Starr CE. Bilateral Salzmann-like nodular corneal degeneration after laser in situ keratomileusis imaged with anterior segment optical coherence tomography and high-frequency ultrasound biomicroscopy. J Cataract Refract Surg. Apr 2009;35(4):785-7.
- Farjo AA, Halperin GI, Syed N, Sutphin JE, Wagoner MD. Salzmann’s nodular corneal degeneration clinical characteristics and surgical outcomes. Cornea. Jan 2006;25(1):11-5.
- Das S, Langenbucher A, Pogorelov P, Link B, Seitz B. Long-term outcome of excimer laser phototherapeutic keratectomy for treatment of Salzmann’s nodular degeneration. J Cataract Refract Surg. Jul 2005;31(7):1386-91.
- Vannas A, Hogan MJ, Wood I. Salzmann’s nodular degeneration of the cornea. Am J Ophthalmol. Feb 1975;79(2):211-9.
- Yoon KC, Park YG. Recurrent Salzmann’s nodular degeneration. Jpn J Ophthalmol. Jul-Aug 2003;47(4):401-4.
- He X, Donaldson KE. Superficial keratectomy for Salzmann nodular degeneration following laser in situ keratomileusis. Can J Ophthalmol. Jun 2019;54(3):e149-e151.
- Qiu J, Cai R, Zhang C. Association between poor wound healing and the formation of Salzmann nodules. J Cataract Refract Surg. Oct 2016;42(10):1527-1530.
- Fong YC, Chuck RS, Stark WJ, McDonnell PJ. Phototherapeutic keratectomy for superficial corneal fibrosis after radial keratotomy. J Cataract Refract Surg. Apr 2000;26(4):616-9.
- Geggel HS. Effect of peripheral subepithelial fibrosis on corneal transplant topography. J Cataract Refract Surg. Jan-Feb 1996;22(1):135-8.
- Roszkowska AM, Spinella R, Aragona P. Recurrence of Salzmann nodular degeneration of the cornea in a Crohn’s disease patient. Int Ophthalmol. Apr 2013;33(2):185-7.
- Papanikolaou T, Goel S, Jayamanne DG, Mudhar H, Desai SP. Familial pattern of Salzmann-type nodular corneal degeneration–a four generation series. Br J Ophthalmol. Nov 2010;94(11):1543.
- Stone DU, Astley RA, Shaver RP, Chodosh J. Histopathology of Salzmann nodular corneal degeneration. Cornea. Feb 2008;27(2):148-51.
- Goerlitz-Jessen MF, Gupta PK, Kim T. Impact of epithelial basement membrane dystrophy and Salzmann nodular degeneration on biometry measurements. J Cataract Refract Surg. Aug 2019;45(8):1119-1123.
- Kim BZ, Wilson PJ, McGhee CN. Annular Salzmann degeneration: Avoiding perturbations and pitfalls in phacoemulsification surgery. J Cataract Refract Surg. Nov 2015;41(11):2580-3.
Faculty Approval By: Brian Zaugg, MD & Griffin Jardine, MD
Copyright: Tanmay Majmudar, ©2023. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Coats Disease Masquerading as Panuveitis
Home / Retina and Vitreous / Other Retinal Vascular Diseases
Title: Coats Disease Masquerading as Panuveitis
Authors: Christopher Le, MSIV University of Colorado School of Medicine; Eric Hansen, MD.
Date: 7/8/2023
Keywords/Main Subjects: coats disease, uveitis masquerade syndromes
Diagnosis: Coats Disease
Description of Case:
Introduction:
Coats disease is a rare, congenital, nonhereditary, idiopathic disorder of the retinal vasculature leading to massive subretinal and intraretinal exudation. The disease typically affects young males in the first or second decades of life and is unilateral.1 The pathophysiology of Coats disease remains unclear – however, it has been proposed that defects in endothelial cells of the retinal vasculature along with abnormal pericytes contribute to the breakdown of the blood-retinal barrier leading to the development of telangiectatic vessels, retinal ischemia, and exudation of lipid-rich fluid.2 Patients with Coats disease have varying symptoms on initial presentation including: decreased visual acuity, eye pain, strabismus, and leukocoria.3 Rarely, Coats disease may present with symptoms and exam findings suggestive of intraocular inflammatory conditions such as uveitis or endophthalmitis. Diseases that present with an intraocular inflammatory picture but are not caused by immune-mediated or infectious processes are referred to as uveitis masquerade syndromes (UMS).4
Case Presentation:
We present a case of Coats disease in a 14-year-old male masquerading as a panuveitis. The patient presented to the emergency department at an outside institution with a three-day history of blurred vision, pain, and redness in his left eye, as well as fever, body aches, and a facial rash. On initial examination, visual acuity in his left eye was markedly decreased at light perception only. There was conjunctival injection and diffuse cell in the anterior chamber and vitreous. On dilated funduscopic examination, a multifocal exudative retinal detachment without macular involvement was appreciated with diffuse subretinal and intraretinal yellow-white exudation and peripheral retinal hemorrhages. Interestingly, the patient reported having a normal dilated eye exam just one year prior.
Shortly after presentation to the outside facility, the patient was transferred to the Moran Eye Center at the University of Utah. Due to concern for possible endophthalmitis or infectious panuveitis, the patient underwent a vitreous and anterior chamber tap as well as injection of broad-spectrum antimicrobials. The patient was evaluated by uveitis specialist who recommended a broad uveitis workup and vitreous biopsy given a dry vitreous tap. The patient’s respiratory viral panel resulted positive for rhinovirus. Broad uveitis workup – including evaluation for HSV, VZV, CMV, toxoplasma, vitreous PCR, quantiferon, RPR, syphilis antibody, bartonella, lysozyme, ACE, and ANCA – returned unremarkable. The patient underwent an exam under anesthesia and a 27-gauge pars plana vitrectomy with vitreous biopsy and intravitreal injections of antimicrobial agents including vancomycin, ceftazidime, clindamycin, and foscarnet. Interestingly, the culture from patient’s vitreous biopsy grew pan-sensitive staphylococcus Capitis. However, the patient’s Karius panel was negative for bacteria and positive for candida. Both positive results were determined to be likely contaminants in consultation with the infectious disease department.
The patient followed-up in clinic and underwent fundus photography and fluorescein angiography following vitrectomy. Fundus examination demonstrated telangiectatic vessels and bulbs with associated diffuse subretinal exudative material and exudative retinal detachment. Fluorescein angiography highlighted the telangiectatic vessels and demonstrated terminal light bulb aneurysms, capillary nonperfusion, and perivascular leakage. The patient was diagnosed with Coats disease, stage 3A given this constellation of findings. The patient was scheduled to undergo laser photocoagulation therapy with adjuvant intravitreal therapy.
At the time of treatment, he was found to have diffuse vitreous hemorrhage obscuring a clear view of the retina with worsening exudative retinal detachment. Subsequently, he was taken to the operating room for a pars plana vitrectomy with scleral buckling, external drainage of exudative material, application of endolaser, silicone oil endotamponade, and intravitreal Avastin injection. At the time of this vitrectomy, the patient’s exudative retinal detachment had worsened to total detachment –classifying the patient’s disease as stage 3B. The patient subsequently underwent multiple exams under anesthesia with laser photocoagulation, sub-tenons Kenalog and intravitreal Avastin injections resulting in resolution of the exudative retinal detachment, significantly improved exudation, and residual fibrosis and vitreoretinal traction sparing the macula. The patient’s visual acuity has improved to counting fingers, and he remains without any signs of neovascular glaucoma.
Discussion:
This case highlights a rare and atypical presentation of Coats disease masquerading as a panuveitis. The critical step in obtaining this patient’s diagnosis, and in diagnosis of Coats disease in general, was obtaining fluorescein angiography with clear view which demonstrated pathognomonic features of the disease: telangiectatic retinal vasculature with terminal light bulb aneurysms, capillary nonperfusion, and perivascular leakage. Coats disease should be included in the differential diagnosis for young males in the first or second decades of life presenting with panuveitis and exudative retinal detachments.
Images or video:
Image 1

Image 1: Fundus photograph taken after initial vitrectomy demonstrating telangiectatic retinal vasculature, yellow subretinal exudative material, and multifocal exudative retinal detachment.
Image 2
Image 2: Widefield fluorescein angiography taken after initial vitrectomy demonstrating telangiectatic vessels with terminal light bulb aneurysms, capillary nonperfusion, and perivascular leakage.
Image 3

Image 3: Fundus photograph taken during most recent exam under anesthesia in June 2023, following several laser photocoagulation and intravitreal Avastin treatments, demonstrating improved exudative retinal detachment, persistent but improved exudation, and residual fibrosis and traction involving the macula.
Summary of the Case:
A 14-year-old male presented with a marked decrease in visual acuity to light perception only, eye pain, and eye redness of the left eye in the setting of fever, chills, and a facial rash. Initial examination was remarkable for a multifocal exudative retinal detachment and significant anterior chamber and vitreous cell suggestive of a panuveitis or intraocular inflammatory condition. Vitreous culture from intraoperative biopsy grew staphylococcus Capitis, but was ultimately determined to be a contaminant. All other elements of the patient’s broad uveitis workup were unremarkable. Fluorescein angiography was performed following the patient’s initial vitrectomy and revealed telangiectatic vessels, capillary nonperfusion, perivascular leakage – findings pathognomonic for Coats disease. The patient underwent a series of interventions including vitrectomy/buckle with external drainage of exudates and several treatments with laser photocoagulation and intravitreal Avastin injections. His most recent examination and fundus photos demonstrated improvement in the exudative retinal detachment and persistent, but improved, exudation. His visual acuity has improved to counting fingers and he remains without any signs of development of neovascular glaucoma.
Format: Case Report
References
-
- Yousef YA, ElRimawi AH, Nazzal RM, et al. Coats’ disease: characteristics, management, outcome, and scleral external drainage with anterior chamber maintainer for stage 3b disease. Medicine (United States). 2020;99(16):E19623. doi:10.1097/MD.0000000000019623
- Sen M, Shields CL, Honavar SG, Shields JA. Coats disease: An overview of classification, management and outcomes. BMC Ophthalmol. 2017;17(1):1. doi:10.4103/ijo.IJO
- Yang X, Wang C, Su G. Recent advances in the diagnosis and treatment of Coats’ disease. Int Ophthalmol. 2019;39(4):957-970. doi:10.1007/s10792-019-01095-8
- Hsu YR, Wang LU, Chen FT, et al. Clinical Manifestations and Implications of Nonneoplastic Uveitis Masquerade Syndrome. Am J Ophthalmol. 2022;238(August 2020):75-85. doi:10.1016/j.ajo.2021.12.018
Faculty Approval by: Eric Hansen, MD; Griffin Jardine, MD.
Copyright: Christopher Le, © 2023. For further information regarding the rights to this collection, please visit: http://morancore.utah.edu/terms-of-use/
Identifier: Moran_CORE_126934