


Retinal Vasculitis
Home / Ophthalmic Pathology and Intraocular Tumors / Retina and Retinal Pigment Epithelium
Title: Retinal Vasculitis Case Report
Author: Sherief Raouf, Visiting Medical Student from the Stony Brook School of Medicine
Photographer: Glen Jenkins
Date: Friday, August 19, 2016
Image:


Diagnosis: Idiopathic mixed arterial/venous occlusive vasculitis
Secondary CORE Category: Retina and Vitreous / Focal and Diffuse Choroidal and Retinal Inflammation
Description of Image:
| Negative Laboratory Workup | |
| ANCA | Systemic necrotizing vasculitis |
| Serine Proteinase Ab | Systemic vasculitis |
| HbSAg, HbSAb | Hep B |
| ANA | Sensitive for SLE |
| Anti-Smith Ab | Specific for SLE |
| dsDNA Ab | Lupus Nephritis & SLE |
| Rh Factor | Rheumatoid Arthritis |
| Cardiolipin Ab and B2GP Ab | Anti-phospholipid syndrome |
| SSA Ab and SSB Ab | Sjogren’s & SLE |
| Ribonucleic Protein Ab | SLE & Mixed connective tissue disease |
| SCL-70 Ab | Scleroderma |
| PT, PTT, dRVVT | Hypercoagulability |
| Table 1. | |
Background: The patient is a 52 year old woman with a past medical history of diabetes mellitus and hypothyroidism who presented complaining of reduced vision OD, described as a “large grey spot in the center.” Visual acuity was 20/30 OD, 20/20 OS, pupils equal and reactive to light with no afferent pupillary defect, and the anterior chamber deep and quiet OU. The vitreous OD demonstrated +1 cells and the macula OD exhibited pigmentary atrophy and a small superior branch retinal artery occlusion (BRAO), while both eyes showed signs of periphlebitis.
There were no obvious signs of systemic inflammatory disease including Lupus, Sjogren’s, Giant-cell Arteritis, Granulomatosis with polyangiitis, or other systemic vasculitis. The review of systems was negative for cough, fever, orogenital ulcerations (Behçet disease), hearing loss and encephalopathy (Susac syndrome), or skin rash. Initial laboratory workup is shown in Table 1.
A laboratory investigation of infectious causes was performed and the following tests were negative: quantiferon, FTA-ABS, RPR, B. Burgdorferi Ab, toxoplasmosis and HIV ELISA. A laboratory workup for hypercoagulability was also negative (including PT, PTT, dRVVT). Brain MRI was negative for any acute intracranial process or findings of vascular inflammation or stenosis. Chest X-ray showed normal vascular markings, and a lack of any nodules, consolidations or hilar adenopathy.
The diagnosis of an arterial occlusive vasculitis was made, and treatment with oral Prednisolone and Mycophenolate mofetil was begun. One month later, the patient returned complaining of worsening blurriness OD. Exam revealed decreased vitreous cells with a persistent superior BRAO, periphlebitis and retinal neovascularization OD. The decision to treat the right eye with peripheral panretinal photocoagulation (PRP) was made. Over the ensuing months, the retinal vasculitis was medically managed with courses of prednisolone, mycophenolate and methotrexate and regular follow-up maintained. The photos above are taken from a visit 12 months after the initial visit, upon which a new BRAO was discovered.
The fundus photo demonstrates the gray-white sheathing of a retinal branch artery that is characteristic of a retinal vasculitis.1 The perivascular sheathing in occlusive vasculitis is thought to be an exudate of inflammatory cells around the vessel that leads to occlusion (Figure A, yellow arrow). Occlusion of the retinal vessel can result in ischemia of the retina and areas of capillary non-perfusion. This fundus photo demonstrates such an area of ischemic retina that is seen downstream and superior to the occluded vessel. This retina also exhibits neovascularization coincident with the ischemic areas of retina.
The late frame fluorescein angiography shows evidence of vascular obstruction in the inferior arcade, distal to the sheathed retinal branch artery. There is evidence of diffuse retinal vasculitis and vessel leakage. In addition, we can see the area of prior PRP of the areas of retinal capillary non-perfusion (Figure B, blue arrow). The angiography is able to additionally define the zones where the loss of retinal perfusion (Figure A, white arrow) has led to new areas of leaking neovascularization. Notably, there is a pronounced macular hyperfluorescence that corresponds to neovascularization (Figure B, green arrow). Given that neovascularization has continued, and that the vasculitis is active, the decision to restart corticosteroid treatment was undertaken with a plan for another round of PRP. Typically it would be preferable to ensure that the vasculitis is inactive when PRP is implemented, as its use can result in the release of more angiogenic factors, aggravating neovascularization.2
A key distinction to be made in making this diagnosis is between that of primary branch retinal vein occlusions, which closely mimics idiopathic retinal vasculitis. In BRVO, the occlusions typically occur at arteriovenous crossings, and are not multiple nor as peripheral as they tend to be in vasculitic obstructions.3 Finally, it should be said that there is some consensus to label idiopathic retinal vasculitis as Eales disease when vascular occlusions and neovascularization lead to recurrent vitreous hemorrhage in the presence of serological evidence of tuberculosis.3 Admittedly, this distinction may be a semantic one, as the yet unsolved pathophysiology may one day reveal these two entities to lie on one spectrum.
The differential for retinal vasculature occlusions is very broad and can be usefully divided into non-inflammatory (Diabetes, BRVO) and inflammatory causes. Inflammatory etiologies were examined with a careful history and an extensive laboratory and radiologic workup, yielding no clear cause. Indeed, many inflammatory retinal vascular obstructions are secondary to a systemic inflammatory process (infectious and non-infectious). However, primary idiopathic retinal vasculitis often is isolated to the eye and absent of any systemic involvement, frequently making this a diagnosis of exclusion.4
Format: Fundus photography and fluorescein angiography
References:
- Gass, J. Donald M. Stereoscopic Atlas of Macular Diseases: Diagnosis and Treatment. St. Louis: Mosby, 1997. Print.
- Biswas, J. et al. Eales disease–an update. Surv Ophthalmol 47, 197–214 (2002).
- Namperumalsamy, P., and Dhananjay S. “Eales ” Retina. Stephen Ryan MD et al. 5th ed. Oxford: Saunders, 2013. 1479-1485. Print.
- Saurabh, K., Das, R., Biswas, J. & Kumar, A. Profile of retinal vasculitis in a tertiary eye care center in Eastern India. Indian Journal of Ophthalmology 59, 297 (2011).
Faculty Approval By: Dr. Akbar Shakoor, Dr. Griffin Jardine
Identifier: Moran_CORE_23842
Disclosure: No financial disclosures to share
Fundus Photography and Fluorescein Angiography of Familial Exudative Vitreoretinopathy
Home / Pediatric Ophthalmology and Strabismus / Disorders of the Retina and Vitreous
Title: Fundus Photography and Fluorescein Angiography of Familial Exudative Vitreoretinopathy
Author: Kenneth Price, BS
Photographer: Unknown
Date: 7/29/2016
Image or video:


Keywords/Main Subjects: Familial Exudative Vitreoretinopathy; FEVR
Secondary CORE Category: Retina and Vitreous / Congenital and Developmental Abnormalities
Diagnosis: Familial Exudative Vitreoretinopathy
Description of Image: Familial Exudative Vitreoretinopathy (FEVR) is a rare inherited disorder of retinal blood vessel development which leads to incomplete vascularization of the peripheral retina. Inheritance can be autosomal dominant, recessive, X-linked, or sporadic. The disease ranges from asymptomatic to severe. If there is sufficient retinal ischemia secondary vascular proliferation can lead to fibrosis, traction, retinal detachment and retinal dysplasia. FEVR needs to be distinguished from ROP due to their similar appearances. The diagnosis of FEVR is made in patients who were born at full term or otherwise have findings inconsistent with ROP and can further be ruled in by genetic testing, specifically testing for FZD4, LRP5, TSPAN12, NDP and FZ mutations among others. Many patients diagnosed with FEVR retain vision of 20/40 or better. Macular ectopia, retinal folds, and retinal detachments are the main causes for visual loss. A fundamental component of diagnosis and treatment is identifying peripheral retinal areas of non-perfusion by performing fluorescein angiography, often under anesthesia due to this disease most commonly presenting in the pediatric age range. Wide-field angiography has become particularly useful in this disease.
This fundus photo and fluorescein angiography photo are from a 4 year old female who was diagnosed with FEVR at an early age after her parents began to notice symptoms of vision loss. On exam, she was found to have an avascular retina peripherally and was followed until she developed neovascularization and associated fibrosis. Fundus photography and fluorescein angiography here show a broad linear macular fold with associated epiretinal membrane and peripheral avascular retina with areas of photocoagulation. She was treated with peripheral photocoagulation, vitrectomies to release vitreoretinal traction, and a scleral buckle for retinal detachment.
References:
Gilmour DF. Familial exudative vitreoretinopathy and related retinopathies. Eye (2015) 29, 1–14.
John VJ, McClintic JI, Hess DJ, Berrocal AM. Retinopathy of Prematurity Versus Familial Exudative Vitreoretinopathy: Report on Clinical and Angiographic Findings. Ophthalmic Surg Lasers Imaging Retina. 2016 Jan;47(1):14-9.
Faculty Approval by: M.E. Hartnett; Griffin Jardine
Disclosure (Financial or other): None
Copyright statement: Copyright 2016. Please see terms of use page for more information.
Stargardt Disease
Home / Retina and Vitreous / Hereditary Retinal and Choroidal Dystrophies
Title: Stargardt disease
Author (s): Jamie Odden, MS4 MPH
Photographer: unknown
Date: 8/16/2016
Image or video:

Figure 1: “Beaten bronze” central macular atrophy surrounded by yellow, round pisciform flecks.

Figure 2: Fundus autofluorescence demonstrates a hypofluorescent macula corresponding to atrophy and surrounding hyperfluorescent spots corresponding to lipofuscin deposits in the RPE.

Figure 3: OCT. Thinning and disorganization of the inner segment-outer segment (IS-OS) junction of the photoreceptors in the macula (yellow arrow). Accentuated choroidal reflectivity (green bar).
Keywords/Main Subjects: Stargardt Disease; Central macular atrophy; Inherited macular dystrophy
Diagnosis: Stargardt Disease
Description of Image:
Epidemiology: Stargardt disease is the most common inherited macular dystrophy, with a prevalence of approximately 1 in 8,000-10,000 individuals. It is a common cause of central vision loss in individuals under 50 years old, with typical onset between 10-20 years old.
Genetics: The underlying etiology is due to accumulation of lipofuscin in the retinal pigment epithelium (RPE). Most cases are autosomal recessive due to mutations in the ABCA4 gene, which encodes for a transporter protein expressed by rod outer segments. ABCA4 mutations can cause toxins to accumulate in the photoreceptors, leading to formation of lipofuscin. Fewer cases are autosomal dominant due to a mutation in ELOVL4, which encodes a component of the fatty acid elongation system in photoreceptors.
Clinical exam: Classic fundoscopic findings include “beaten bronze” central macular atrophy surrounded by yellow round or pisciform flecks (Figure 1). The condition is referred to as “fundus flavimaculatus” if the discrete yellow flecks are widespread throughout the fundus retinal pigment epithelium (RPE). “Fundus flavimaculatus” is a milder condition rendering better visual function due to less macula involvement.
Diagnosis/testing: Fundus autofluorescence (FAF) and optical coherence tomography (OCT) can confirm diagnosis and help stage the disease. FAF and OCT often detect RPE changes before they are found on clinical fundoscopic exam. Commonly, FAF shows a hypofluorescent macula, corresponding to atrophy, and surrounding hyperfluorescent spots, corresponding to lipofuscin deposits in the RPE (Figure 2).
OCT demonstrates thinning and disorganization of the inner segment-outer segment (IS-OS) junction of the photoreceptors in the macula (Figure 3). Total loss of the IS-OS is possible over time. Choroidal hyper-reflectivity often occurs due to overlying retinal atrophy.
Historically, the “dark choroid sign” on fluorescein angiography (FA) was used to confirm clinical diagnosis, though FA has been largely replaced by FAF and OCT. The sign is present in over 80% of patients. Evidence suggests that the sign is caused by accumulation of lipofuscin throughout the RPE which masks background choroidal fluorescence.
Clinical course: In most cases, central vision loss is slow and progressive. Later-onset disease is associated with a better prognosis. Visual acuity ranges from 20/50 to 20/200, with most individuals maintaining fair acuity in one eye.
Management: Stargardt disease is incurable. No treatments are available to slow progression, though pharmacologic and genetic therapies are under investigation. Individuals should avoid vitamin A supplementation and minimize exposure to bright sunlight. Additionally, low vision therapy should be considered.
Format: image
References:
- North V, Gelman R, Tsang SH. Juvenile-Onset Macular Degeneration and Allied Disorders. Developments in ophthalmology. 2014;53:44-52. doi:10.1159/000357293.
- Liu A, Lin Y, Terry R, Nelson K, Bernstein PS. Role of long-chain and very-long-chain polyunsaturated fatty acids in macular degenerations and dystrophies. Clin Lipidol. 2011;6(5):593-613.
- Regillo C, ed. Basic and clinical science course (BCSC) 2012-2013: Retina and vitreous section 12. San Francisco, United States: American Academy of Ophthalmology; 2012.
Identifier: Moran_CORE_23820
Faculty Approval by: Paul Bernstein, MD PhD; Griffin Jardine, MD
Disclosure (Financial or other): None
Copyright statement: Copyright 2015. Please see terms of use page for more information.
Primary Acquired Melanosis (PAM) with Atypia: Pathology and Clinical Correlations
Home / External Disease and Cornea / Neoplasms of the Conjunctiva and Cornea
Title: Primary Acquired Melanosis (PAM) with Atypia: Pathology and Clinical Correlations
Author (s): Jack Li, BA
Photographer:
- Pathology Photomicrographs: Jack Li
- Clinical Photo: Dr. Amy Lin
Date: 9/12/2016
Image:

Figure A: External photography of the eye in a patient with primary acquired melanosis. Diffuse melanosis of varying degrees of pigmentation noted.

Figure B: The same eye after surgical excision and cryotherapy. The melanotic lesions have been removed.

Figure C: Medium magnification H&E stain of the conjunctiva. Stratified squamous non-keratinized epithelium is observed. Melanin-laden cells are noted near the basement membrane. Atypical cells are appreciated clustered near the basement membrane.

Figure D: Mid-high magnification of H&E stain of the conjunctiva. Atypical cells can be appreciated mid-way through the epithelium. However, atypical cells have not invaded the entire depth of the epithelium.
Secondary CORE Category: Ophthalmic Pathology / Conjunctiva
Keywords/Main Subjects: Primary Acquired Melanosis, Melanocytic Lesions of Ocular
Diagnosis: Primary Acquired Melanosis with Atypia
Description of Images:
Primary acquired melanosis (PAM) of the conjunctiva is a pigmented lesion of the conjunctiva that is flat, painless and non-cystic. PAM typically occurs unilaterally, is more likely to occur in lightly pigmented individuals and is most likely to present in the 6th decade of life. PAM represents 11% of all conjunctival tumors and 21% of all conjunctival melanocytic lesions1. Figure A is the external photography of a 60-year-old female with approximately 13-year history of PAM of the conjunctiva. Features associated with PAM include unilaterality, waxing and waning of size and pigment over time and a mottled or dusted pigmented appearance. PAM most commonly occurs on the bulbar conjunctiva, limbal conjunctiva and cornea. The typical patient is a Caucasian adult presenting around 60 years of age. PAM is divided histopathologically into PAM with or without atypia. PAM with atypia has a high chance of progression into melanoma while PAM without atypia has little chance of progressing to melanoma2. Biopsy and histopathologic examination allow determination of the presence or absence of atypia. Because PAM with atypia has significant risk of progression into melanoma, a potentially lethal tumor, surgical and medical intervention is warranted. Figure B represents the same patient after excision of the lesion and cryotherapy.
PAM without atypia is defined as pigmentation of the conjunctival epithelium with or without benign melanocytic hyperplasia. PAM with atypia is characterized by the presence of atypical melanocytic hyperplasia. Mild atypia is defined as atypical melanocytes confined to the basal layer of the epithelium; severe atypia is defined as atypical melanocytes that extend into the superficial non-basal portion of the epithelium and may contain epitheloid cells1. Figure C and D are low and medium H&E photomicrographs of the specimen obtained from the patient where pigmentation along the basal layer of the epithelium can be appreciated. One can appreciate the extension of atypical melanocytes mid-way into the epithelium, qualifying this lesion to be PAM with severe atypia.
Atypia is determined by cytological features and growth patterns that are associated with malignant potential. Four types of atypical melanocytes include the small polyhedral cells, epitheloid cells, dendritic cells, and spindle cells. Polyhedral cells contain small, round nuclei with little cytoplasm. Epitheloid cells contain abundant eosinophilic cytoplasm. Spindle cells are aligned such that the long axis are parallel to the basement membrane. Dendritic cells are large cells with complex branching dendrites found along the basilar layer3.
The mainstay of treatment of PAM with atypia is wide excision of the lesion and cryotherapy of the borders of the lesions. Amniotic graft can be applied to the surgical site to facilitate healing. Topical chemotherapy, most commonly with mitomycin-c, can be used as adjuvant therapy in cases of diffuse lesion, positive surgical margins, large lesions that cannot be completely removed, or in cases of recurrent disease2. Interferon a-2b has shown promise as a medical management. A six-week trial showed that successive application of interferon a-2b resulted in shrinking of PAM4.
References:
- Shields, J. A. et al. Primary acquired melanosis of the conjunctiva: experience with 311 eyes. Trans. Am. Ophthalmol. Soc. 105, 61-71–2 (2007).
- Oellers, P. & Karp, C. L. Management of pigmented conjunctival lesions. Ocul. Surf. 10, 251–263 (2012).
- Folberg, R., McLean, I. & Zimmerman, L. Primary acquired melanosis of the conjunctiva. Hum. Pathol. 16, 129–35 (1985).
- Garip, A. et al. Evaluation of a short-term topical interferon α-2b treatment for histologically proven melanoma and primary acquired melanosis with atypia. Orbit 35, 29–34 (2016).
Faculty Approval by: Amy Lin, MD; Griffin Jardine, MD
Identifier: Moran_CORE_23807
Disclosure (Financial or other): None
Copyright statement: Copyright 2017. Please see terms of use page for more information.
Episcleritis Associated with Lyme Disease
Home / External Disease and Cornea / Diagnosis and Management of Immune-Related Disorders of the External Eye
Title: Episcleritis Associated with Lyme Disease, Case Report
Author: Eliza Barnwell, MSCR
Photographer: unknown
Date: 9/19/16
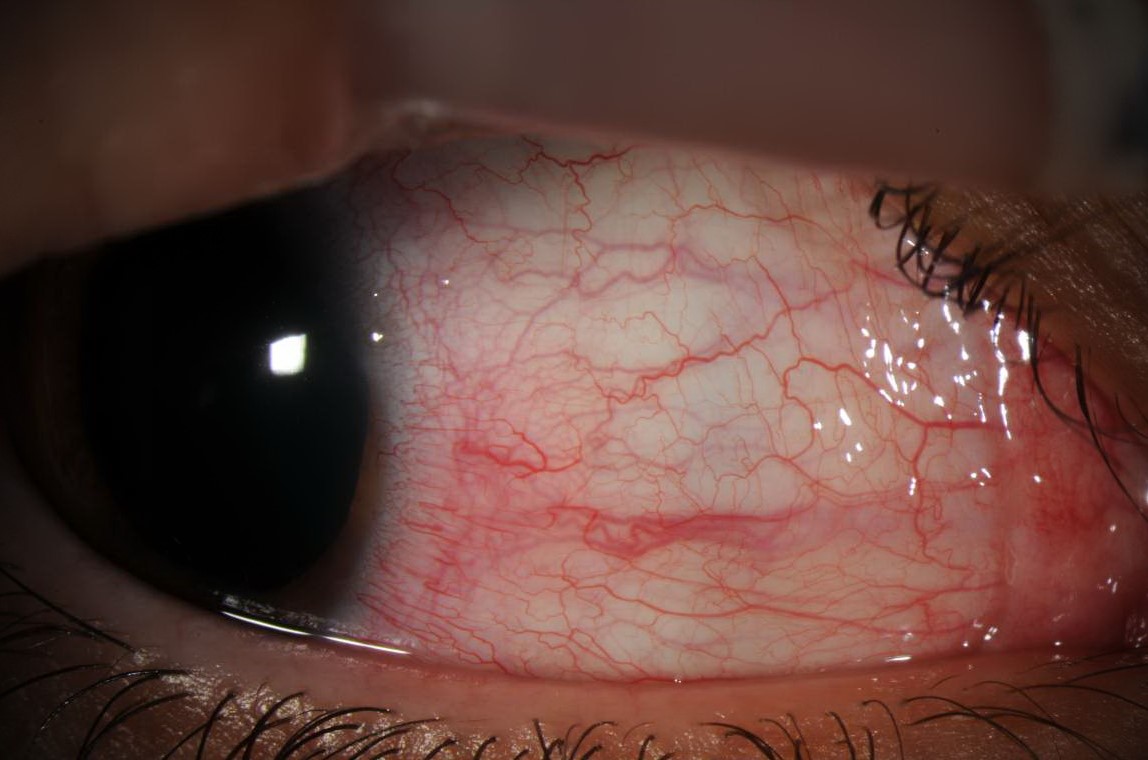
Slit-lamp photograph showing dilated and injected episcleral vessels.

Same patient 5 minutes post-application of 2.5% phenylephrine.
Secondary CORE Category: Intraocular Inflammation and Uveitis / Noninfections (Autoimmune) Uveitis
Keywords / Main Subjects: Episcleritis; scleritis; conjunctival inflammation
Diagnosis: Episcleritis
Description of Image: This patient is a 24–year-old male with a one year history of Lyme disease who presented with 2 months of light sensitivity, redness and irritation in the right eye. Slit-lamp examination of the right eye revealed mild ptosis, 2+ sectoral injection of the conjunctiva in the superior and nasal regions with dilated episcleral vessels and 3 cells per high-powered-field in the anterior chamber. Examination of his left eye was normal. Topical application of 2.5% phenylephrine blanched the injected vessels. He was started on prednisolone drops 4 times a day and the episcleritis resolved shortly thereafter.
Episcleritis is typically an idiopathic, autoimmune condition causing dilation and inflammation of the superficial layers of the eye.1 Although usually idiopathic, episcleritis can also be associated with a variety of systemic diseases such as rheumatoid arthritis, Systemic Lupus Erythematous (SLE) and Inflammatory bowel diseases (IBD).3 Though uncommon, a number of studies have described episcleritis as an ocular manifestation of Lyme disease, both in late and early stages of the disease.4,5,6 In one case report, initiation of intravenous ceftriaxone therapy aggravated episcleral inflammation and caused an anterior chamber reaction in a patient with positive serum titers for Lyme.7
Scleritis is a deeper inflammation of the scleral vessels that typically does not blanch with application of topical phenylephrine. It is often associated with a systemic autoimmune disease in contrast to episcleritis. Scleritis tends to have dramatic focal tenderness and in its more severe form can lead to scleral thinning and perforation.
Treatment for scleritis commonly involves systemic anti-inflammatory and immunosuppressive agents, while episcleritis often resolves on its own or can be treated with a short course of topical steroids versus oral NSAIDs.2
Format: image
Identifier: Moran_CORE_23749
References:
- Jabs DA, Mudun A, Dunn JP, Marsh MJ. “Episcleritis and scleritis: clinical features and treatment results.” American journal of ophthalmology. 2000;130(4):469-76.
- Kirkwood BJ, Kirkwood RA. “Episcleritis and scleritis.” Insight. 2010;35(4):5.
- Akpek EK, Uy HS, Christen W, Gurdal C, Foster CS. “Severity of episcleritis and systemic disease association.” Ophthalmology. 1999;106(4):729-31.
- Flach AJ, Lavoie PE. “Episcleritis, conjunctivitis, and keratitis as ocular manifestations of Lyme disease.” Ophthalmology. 1990;97(8):973-5.
- de la Maza MS, Molina N, Gonzalez-Gonzalez LA, Doctor PP, Tauber J, Foster CS. “Clinical characteristics of a large cohort of patients with scleritis and episcleritis.” Ophthalmology. 2012;119(1):43-50.
- Zaidman GW. “Episcleritis and symblepharon associated with Lyme keratitis. American journal of ophthalmology. 1990;109(4):487-8.
- Mikkilä HO, Seppälä IJ, Viljanen MK, Peltomaa MP, Karma A. “The expanding clinical spectrum of ocular lyme” Ophthalmology. 2000;107(3):581-7.
Faculty Approval By: Dr. Amy Lin, Dr. Severin Pouly and Dr. Griffin Jardine
Orbital Conference: Orbital Inflammation
Home / Orbit, Eyelids, and Lacrimal System / Orbital Inflammatory and Infectious Disorders
 Loading...
Loading...
Title: Orbital Conference: Orbital Inflammation
Authors: Christopher D. Conrady, MD, PhD, Rene Choi, MD, PhD, H. Christian Davidson, MD, and BCK Patel, MD
Date: 06/27/16
Keywords/Main Subjects: orbital cellulitis, infection, inflammation, orbit
Diagnosis/Differential Diagnosis: 2 cases of orbital cellulitis
Brief Description of Case: In the following cases, we present two cases of orbital cellulitis, clinical course, and medical/surgical management. Both patients presented with eyelid swelling. The first patient also had motility deficits upon presentation. They were both found to subsequently have orbital inflammatory events with improvement on IV antibiotics.
Images:
Slide 4: External photograph of patient case 1.
Slide 6-7: CT orbits
Slides 8-12: MRI orbits
Slide 13: CT orbits
Slide 15-18: Pathology slides showing mild, chronic inflammatory cell reaction.
Slide 19: External photograph of patient
Slide 25-26: External photograph of case 2.
Slide 28-32: CT maxillofacial with contrast
Slide 36-40: Pathology of case 2.
Summary of Cases:
These two cases highlight the medical and surgical management of orbital cellulitis.
References for further reading:
- Ebright et al., “Septic Thrombosis of the Cavernous Sinus.” Arch Intern Med, 2001.
- Garcia et al., “Criteria for Nonsurgical Management of Subperiosteal Abscess of the Orbit.” Ophth, 2000
- Harris GJ. Subperiosteal abscess of the orbit. Age as a factor in the bacteriology and response to treatment. Ophthalmology 1994; 101:585-595.
- Harris GJ. Subperiosteal abscess of the orbit: computed tomography and the clinical course. Ophthal Plast Reconstr Surg 1996; 12:1-8.Oxford LE, McClay J. Medical and surgical management of subperiosteal orbital abscess secondary to acute sinusitis in children. Int J Pediatr Otorhinolaryngol 2006; 70:1853-1861.
- Harris, “Subperiosteal Abscess of the Orbit: Age as a Factor in the Bacteriology and Response to Treatment”. Ophth, 2014.
- McKinley et al., “Microbiology of Pediatric Orbital Cellulitis.” AJO, 2007.
- Miranda et al., “Brain abscess: Current management. Neuro Rural Prc, 2013.
- Oxford LE, McClay J. Medical and surgical management of subperiosteal orbital abscess secondary to acute sinusitis in children. Int J Pediatr Otorhinolaryngol 2006; 70:1853-1861.
- Segal et al., “Orbital Complications associated with paranasal sinus infections – A 10-year experience in Israel.” Int J Ped Otorhino, 2016.
- Seltz et al., Microbiology and Antibiotic Management of Orbital Cellulitis. Peds, 2011.
Copyright statement: Christopher D. Conrady, ©2016. For further information regarding the rights to this collection, please visit: URL to copyright information page on Moran CORE
**Signed off on by orbital conference faculty attendees