
Neurofibromatosis
Home / Retina and Vitreous / Pediatric Retina – Posterior Segment Masses
Title: Neurofibromatosis
Authors: Emilie Laura Ungricht, Michael Murri, MD.
Photographer: Dimitrios Malamos, CC BY 4.0 <https://creativecommons.org/licenses/by/4.0>, via Wikimedia Commons
Date: 03/25/2021
Keywords/Main Subjects: Neurofibromatosis
Diagnosis: Neurofibromatosis
Neurofibromatosis is a heritable neurocutaneous disorder involving embryonic neuroectoderm. Neuroectoderm derivatives are the most widely affected, including melanocytes, brain, spinal cord, retina, and parts of the ciliary body of the eye.1, 2 There are two distinct subtypes of neurofibromatosis—neurofibromatosis 1 (NF1) and neurofibromatosis 2 (NF2), each involving different symptoms and with different genetic causes.2,3 Initial signs of neurofibromatosis may present anytime from birth through adolescence and continue to progress through life.3,4
Prevalence
NF1 has an prevalence of 1:2600 to 3000 with approximately half of the cases from familial inheritance and half of the cases from de novo mutations.2, 4-8 NF2 has an incidence of approximately 1:50,000.2,4,7
Genetics
Both NF1 and NF2 are autosomal dominant diseases with high penetrance.2,7 For NF1, the NF-1 gene located on chromosome 17 codes for neurofibromin, a protein that plays a role in cellular growth regulation.2,7 Studies suggest that the mutations affect the Ras pathway.7,9
NF2 involves autosomal dominant mutations in the NF-2 gene, located on chromosome 22. The protein involved is called merlin or schwannomin and is a tumor suppressor involved in key signaling pathways for cell growth and adhesion.10-11
Clinical Diagnostic Criteria and Clinical Features
The diagnostic criteria vary based on the type of neurofibromatosis and the age of the patient. In NF1, diagnosis requires at least 2 of the following criteria: 2,3,12,13
- Skin Findings
- 6 or more café au lait macules, >5mm prepubertal or >15mm post pubertal
- Axillary or inguinal freckles
- 2 or more neurofibromas or 1 plexiform neurofibroma
- Skeletal findings
- Sphenoid dysplasia, tibial pseudoarthrosis or other distinctive bone lesion
- Ocular findings:
- Optic nerve glioma
- 2 or more Lisch nodules (Iris hamartomas)
- Family history of NF1
Lisch nodules (Fig 1) are the most common clinical sign of NF1 and are present in over 94% of patients over the age of 6.14 They are gold to brown hamartomas of the iris that grow up to 2 mm in diameter. These can be visualized with slit-lamp examination.14, 15
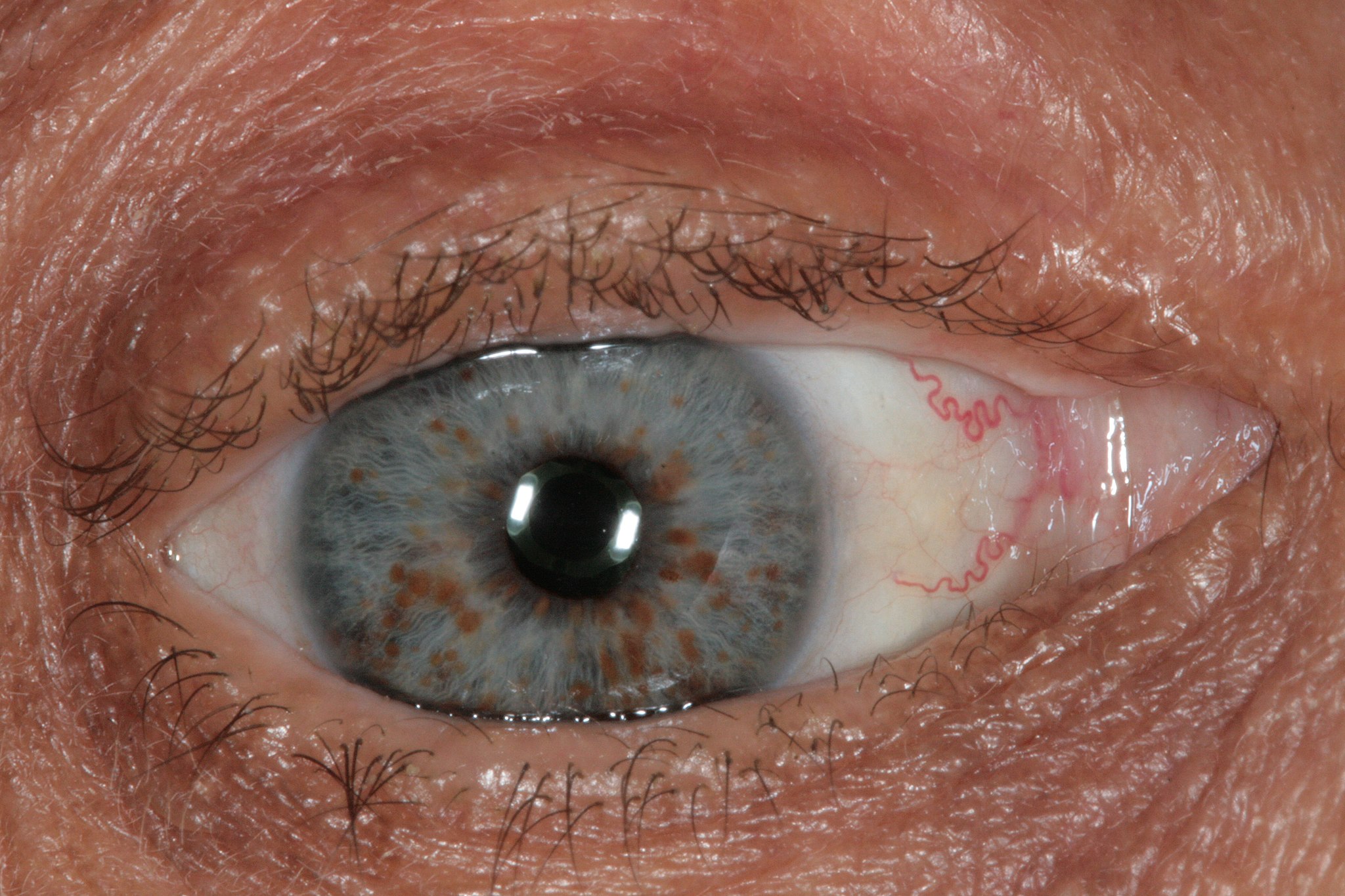
Figure 1: Iris with copper-colored Lisch Nodules, a characteristic finding of NF1. Photo taken by Dimitrios Malamos, CC BY 4.0 <https://creativecommons.org/licenses/by/4.0>, via Wikimedia Commons
Optic nerve gliomas occur in approximately 15-20% of patients with NF1, predominantly developing prior to the age of 7.16-17 Symptoms vary based on the extent of the tumor, but often result in unilateral proptosis, papilledema, and visual field defects.16
NF1 has additional ocular findings that are not included in the diagnostic criteria, notably retinal tumors. These include retinal astrocytic hamartoma, retinal capillary hemangioblastoma, and vasoproliferative tumors.18 These lesions are often located near or on the optic disc. Adult and pediatric patients may develop glaucoma secondary to plexiform neurofibroma infiltrating the angle or from abnormalities of the trabecular meshwork.18
Additional non-ocular findings associated with NF1 include pheochromocytoma and gastrointestinal neurofibromas.19
NF2 is diagnosed with the Manchester Criteria, which requires one of the following:20
- Bilateral vestibular schwannomas in patients <70 years old
- First-degree relative with NF2 and one of the following:
- Unilateral vestibular schwannoma in that relative
- Two of the following: meningioma, cataract, glioma, neurofibroma, schwannoma, posterior subcapsular lenticular opacities
- Unilateral vestibular schwannoma and
- Two of the following: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities
- Multiple meningiomas and one of the following
- Unilateral vestibular schwannoma
- Any two of: schwannoma, glioma, neurofibroma, cataract
Common ocular findings in NF2 include either cortical or posterior subcapsular cataracts. Retinal hamartomas and Lisch nodule can also be present, but are less common than NF1.20 Symptoms often appear in puberty or early adulthood.
Diagnostic Studies
Based on the high rates of ocular involvement in NF1, patients demonstrating any NF1 findings should have a complete ophthalmologic examination. 23 Evaluation of the optic nerve and its function through afferent pupillary testing, color vision, visual fields, dilated exams, and ocular coherence tomography may help to detect optic nerve gliomas. Slit lamp examination can be used to examine the iris for Lisch nodules, optic disc for retinal tumor evaluation. Intraocular pressure measurements and optic nerve evaluation can help detect glaucoma in NF1. CT scan and MRI are useful in the localization of optic gliomas, although these are not always indicated.23,24 Retinal astrocytomas can be detected via fluorescein angiography, demonstrating hypofluorescence in areas of hyperpigmentation.25
Ocular Management
Management depends on the clinical features. Retinal astrocytomas are often monitored over time and are only treated if they progress to retinal detachment or retinal tears. Treatment involves retinal detachment repair, photocoagulation, or cryopexy depending on extent of the pathology.
Optic nerve gliomas often progress slowly and are only treated if they demonstrate rapid growth. If growth is rapid, the glioma can be surgically resected.16 Depending on the extent of the glioma the surgery can be globe preserving or include enucleation. Unfortunately, visual outcomes do not typically improve with glioma removal. Radiation and chemotherapy have been used with some success.26
For glaucoma associated NF1, medical management is typically attempted first. In cases where this fails, surgical intervention has proven to be effective.27
Cataracts in NF2 are typically followed until they become visually significant. They do not often require early surgical intervention.
References:
- Larsen’s Human Embryology(Fifth ed.). Elsevier. 2015. p. 77. ISBN 978-1-4557-0684-6.
- Neurofibromatosis Fact Sheet. (n.d.). Retrieved January 30, 2021, from https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurofibromatosis-Fact-Sheet
- Neurofibromatosis 1. (2020, June 15). Retrieved January 30, 2021, from https://rarediseases.org/rare-diseases/neurofibromatosis-type-1-nf1/
- Nguyen, T.V., Matthews, M.R., Barrera, F.F. and Browning, J.C. (2012), Multiple Cutaneous Plexiform Schwannomas as Initial Presentation of Neurofibromatosis 2 in a 9‐Year‐ Pediatric Dermatology, 29: 536-538. https://doi-org.ezproxy.lib.utah.edu/10.1111/j.1525-1470.2011.01532.x
- Huson, D. Compston, P. Clark, et al. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet, 26 (1989), pp. 704-711
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1.Am J Epidemiol. 2000 Jan 1; 151(1):33-40.
- R. Antonio, E.M. Goloni-Bertollo, L.A. Tridico. Neurofibromatosis: chronological history and current issues. An Bras Dermatol, 88 (2013), pp. 329-343
- E. Ferner, S.M. Huson, N. Thomas, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet, 44 (2007), pp. 81-88
- Esposito, Teresa, Piluso, Giulio, Saracino, Dario, Uccello, Rossella, Schettino, Carla, Dato, Clemente, Capaldo, Guglielmo, Giugliano, Teresa, Varriale, Bruno, Paolisso, Giuseppe, Di Iorio, Giuseppe, and Melone, Mariarosa A. B. “A Novel Diagnostic Method to Detect Truncated Neurofibromin in Neurofibromatosis 1.” Journal of Neurochemistry6 (2015): 1123-128. Web.
- Thaxton, Courtney, Bott, Marga, Walker, Barbara, Sparrow, Nicklaus A, Lambert, Stephen, and Fernandez-Valle, Cristina. “Schwannomin/merlin Promotes Schwann Cell Elongation and Influences Myelin Segment Length.” Molecular and Cellular Neurosciences1 (2011): 1-9. Web.
- SCOLES, Daniel R, NGUYEN, Vu D, Yun Qin, SUN, Chun-Xiao, MORRISON, Helen, GUTMANN, David H, and PULST, Stefan.-M. “Neurofibromatosis 2 (NF2) Tumor Suppressor Schwannomin and Its Interacting Protein HRS Regulate STAT Signaling.” Human Molecular Genetics25 (2002): 3179-189. Web.
- . National Institutes of Health Consensus Development Conference. Neurofibromatosis: conference statement. Arch Neurol 1988;45:575-8.
- Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278:51-7
- E. KingJr, A.C. Zamora. NEUROFIBROMATOSIS, Encyclopedia of Respiratory Medicine, 2006
- John C Nichols, MD; Josh E Amato, MD; Sophia M Chung, MD. Characteristics of Lisch Nodules in Patients With Neurofibromatosis Type 1. Journal of Pediatric Ophthalmology and Strabismus. 2003;40(5):293-296https://doi.org/10.3928/0191-3913-20030901-11
- Cassina M, Frizziero L, Opocher E, et al. Optic Pathway Glioma in Type 1 Neurofibromatosis: Review of Its Pathogenesis, Diagnostic Assessment, and Treatment Recommendations. Cancers (Basel). 2019;11(11):1790. Published 2019 Nov 14. doi:10.3390/cancers11111790
- Trevisson E, Cassina M, Opocher E, Vicenzi V, Lucchetta M, Parrozzani R, Miglionico G, Mardari R, Viscardi E, Midena E, Clementi M. Natural history of optic pathway gliomas in a cohort of unselected patients affected by Neurofibromatosis 1. J Neurooncol. 2017 Sep; 134(2):279-287.
- Destro M, D’Amico DJ, Gragoudas ES, Brockhurst RJ, Pinnolis MK, Albert DM, Topping TM, Puliafito CA. Retinal manifestations of neurofibromatosis. Diagnosis and management. Arch Ophthalmol. 1991 May;109(5):662-6. doi: 10.1001/archopht.1991.01080050076033. PMID: 1902661.
- FRED H. HOCHBERG, AMAURI BATISTA DASILVA, JAMES GALDABINI, E. P. RICHARDSON. Gastrointestinal involvement in von Recklinghausen’s neurofibromatosis. Neurology Dec 1974, 24 (12) 1144; DOI: 10.1212/WNL.24.12.1144
- Evans DG, King AT, Bowers NL, Tobi S, Wallace AJ, Perry M, Anup R, Lloyd SKL, Rutherford SA, Hammerbeck-Ward C, Pathmanaban ON, Stapleton E, Freeman SR, Kellett M, Halliday D, Parry A, Gair JJ, Axon P, Laitt R, Thomas O, Afridi S, Ferner RE, Harkness EF, Smith MJ; English Specialist NF2 Research Group. Identifying the deficiencies of current diagnostic criteria for neurofibromatosis 2 using databases of 2777 individuals with molecular testing. Genet Med. 2019 Jul;21(7):1525-1533. doi: 10.1038/s41436-018-0384-y. Epub 2018 Dec 7. PMID: 30523344.
- Ragge, Nicola K, Baser, Michael E, Klein, Jana, Nechiporuk, Alex, Sainz, Jesus, Pulst, Stefan-M, and Riccardi, Vincent M. “Ocular Abnormalities in Neurofibromatosis 2.” American Journal of Ophthalmology5 (1995): 634-41. Web.
- Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009 Jun 19;4:16. doi: 10.1186/1750-1172-4-16. PMID: 19545378; PMCID: PMC2708144.
- Miller DT, Freedenberg D, Schorry E, Ullrich NJ, Viskochil D, Korf BR; COUNCIL ON GENETICS; AMERICAN COLLEGE OF MEDICAL GENETICS AND GENOMICS. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics. 2019 May;143(5):e20190660. doi: 10.1542/peds.2019-0660. PMID: 31010905.
- Stewart DR, Korf BR, Nathanson KL, Stevenson DA, Yohay K. Care of adults with neurofibromatosis type 1: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018 Jul;20(7):671-682. doi: 10.1038/gim.2018.28. Epub 2018 Apr 26. PMID: 30006586.
- Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13:834–843. [PubMed] [Google Scholar]
- Tsang DS, Murphy ES, Merchant TE. Radiation Therapy for Optic Pathway and Hypothalamic Low-Grade Gliomas in Children. Int J Radiat Oncol Biol Phys. 2017 Nov 1;99(3):642-651. doi: 10.1016/j.ijrobp.2017.07.023. Epub 2017 Jul 23. PMID: 29280458.
- Thavikulwat, AT, Edward, DP, AlDarrab, A, Vajaranant, TS. Pathophysiology and management of glaucoma associated with phakomatoses. J Neuro Res. 2019; 97: 57– 69. https://doi.org/10.1002/jnr.24241
Identifier: Moran_CORE_67595
Copyright statement: Copyright 2021. Please see terms of use page for more information.