
Choroideremia
Home / Retina and Vitreous / Hereditary Retinal and Choroidal Dystrophies
Title: Choroideremia
Author (s): Liang Cheng, 4th Year Medical Student, University of Iowa
Photographer: Unknown
Date: 06/20/2017
Images:

Keywords/Main Subjects: Choroideremia
Diagnosis: Choroideremia
Description of Image:
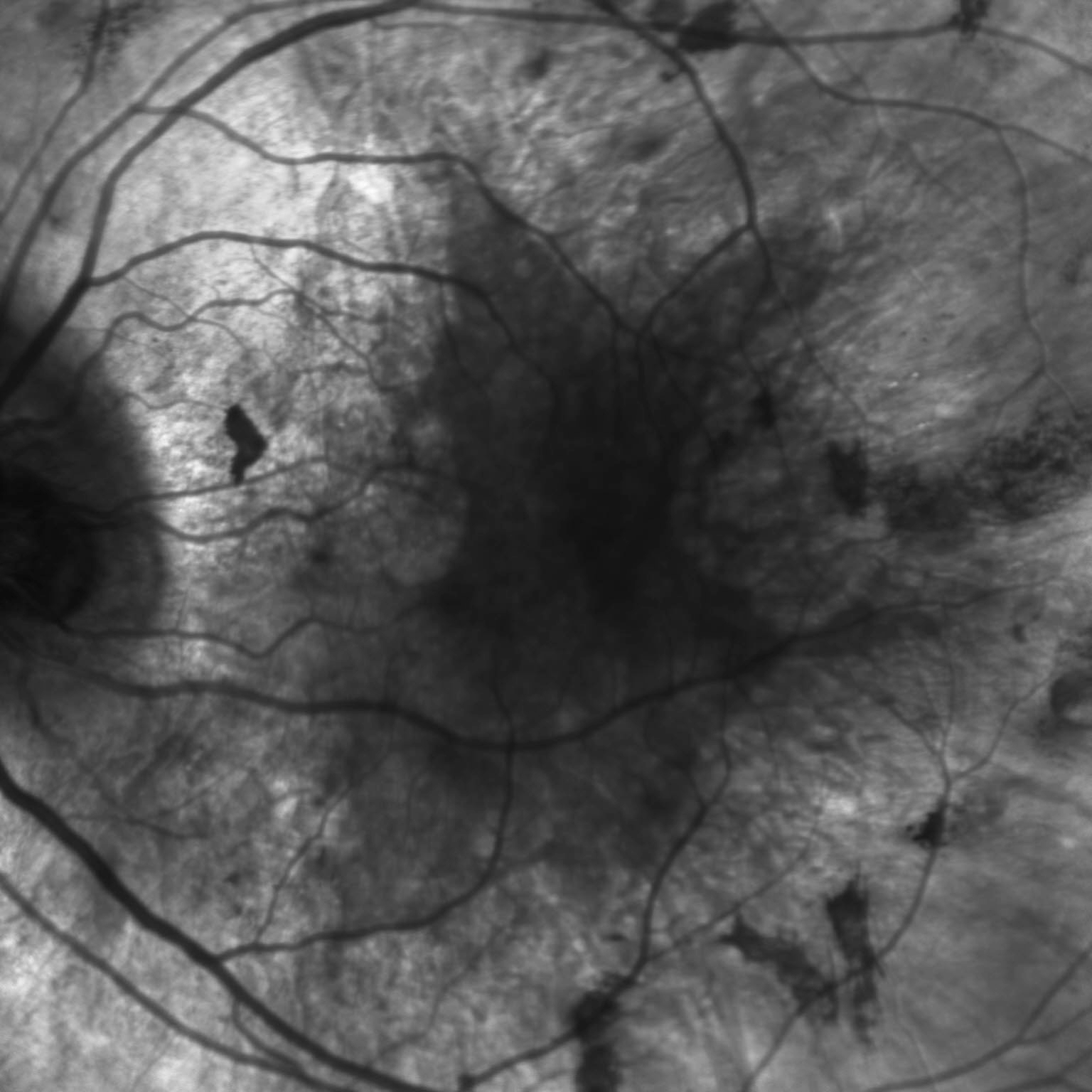
Choroideremia is a rare X-linked retinal degeneration that typically affects males. It is characterized by progressive degeneration of the retinal pigment epithelium, photoreceptors, and choriocapillaris. In fact, in late stages, the underlying sclera is exposed due to complete chorioretinal dystrophy, hence the name choroid-“eremia” (Greek for bare). The disease is caused by a mutation of the CHM gene, which leads to defects in intracellular vesicular trafficking and subsequent retinal pigment epithelium (RPE), photoreceptor, and choroidal degeneration.
Patients typically present with night blindness and peripheral vision loss in teenage years. Central vision is maintained until the 5th or 6th decade of life but then can rapidly deteriorate. Carriers are mostly unaffected, but there are case reports of symptomatic female patients with less severe features. On dilated fundus exam, the earliest sign is diffuse pigment clumping, followed by atrophy with visible sclera and choroidal vessels. Atrophy progresses centripetally and the fovea is the last to become affected. Choroideremia is usually suspected based on the symptoms and findings, but a full clinical work-up includes a detailed family history, visual field testing, electroretinography (ERG), and genetic testing. Visual field testing shows patchy peripheral vision loss that corresponds to the location of the chorioretinal dystrophy, and ERG shows a reduced scotopic component early on in the disease process. Additional imaging can also be supportive of the diagnosis, such as fundus autofluorescence, infrared (IR) and optical coherence tomography. There are currently no treatments available, but gene therapy trials are underway. The differential to be considered includes retinitis pigmentosa, gyrate atrophy, and ocular albinism.
These color and IR fundus images are of a 35 year old gentleman with an established family history of choroideremia who was diagnosed with the disease in his late 20’s. The color fundus photo of his left eye shows scattered pigment clumping and diffuse atrophy. One can appreciate the bare sclera around the scalloped edge of the remaining healthy retina, as well as the visible large choroidal vessels. The IR fundus photo confirms the retinal atrophy in the periphery with the scalloped border. These findings support patient’s complaints of poor peripheral vision and trouble with night-time driving.
References:
Sorsby A, Franceschetti A, Joseph R, Davey JB. Choroideremia; clinical and genetic aspects. The British journal of ophthalmology. 1952;36(10):547-581.
Huckfeldt RM, Bennett J. Promising first steps in gene therapy for choroideremia. Human gene therapy. 2014;25(2):96-97.
Morgan JI, Han G, Klinman E, et al. High-resolution adaptive optics retinal imaging of cellular structure in choroideremia. Investigative ophthalmology & visual science. 2014;55(10):6381-6397.
Bonilha VL, Trzupek KM, Li Y, et al. Choroideremia: analysis of the retina from a female symptomatic carrier. Ophthalmic genetics. 2008;29(3):99-110.
Faculty Approval by: Dr. Bernstein, Griffin Jardine MD
Identifier: Moran_CORE_24177
Copyright statement: Copyright 2017. Please see terms of use page for more information.