
Uvea
Home / Ophthalmic Pathology / Uveal Tract
Uveal Tract


Normal globe


Normal iris and ciliary body
Uveal tract consists of the iris, ciliary body, and choroid. It is embryologically derived from neuroectodermand neural crest
Firm attachments exist at 3 sites:
- Scleral spur
- Exit points at vortex veins
- Optic nerve
Iris


Low magnification view of normal iris


Higher magnification view of normal iris
The iris separates the anterior segment into anterior +posterior chambers
- The iris is made up of 5 layers:
- Anterior border layer–condensation ofiris stroma + melanocytes
- Stroma–blood vessels, nerves, melanocytes, fibrocytes, and clump cells
- Muscular layer–dilator + sphincter muscle
- Anterior pigment epithelium
- Posterior pigment epithelium
- Clump cells
- Type I (clump cells of Koganei): macrophages
- Type II:smooth muscle cells
- Stromal vessels are surrounded with collagen fibrils and appear thick walled
- Color of the eye determined by size + number of melanin granules in anterior stroma
- Cell bodies of anterior pigment layer give rise to dilator muscle
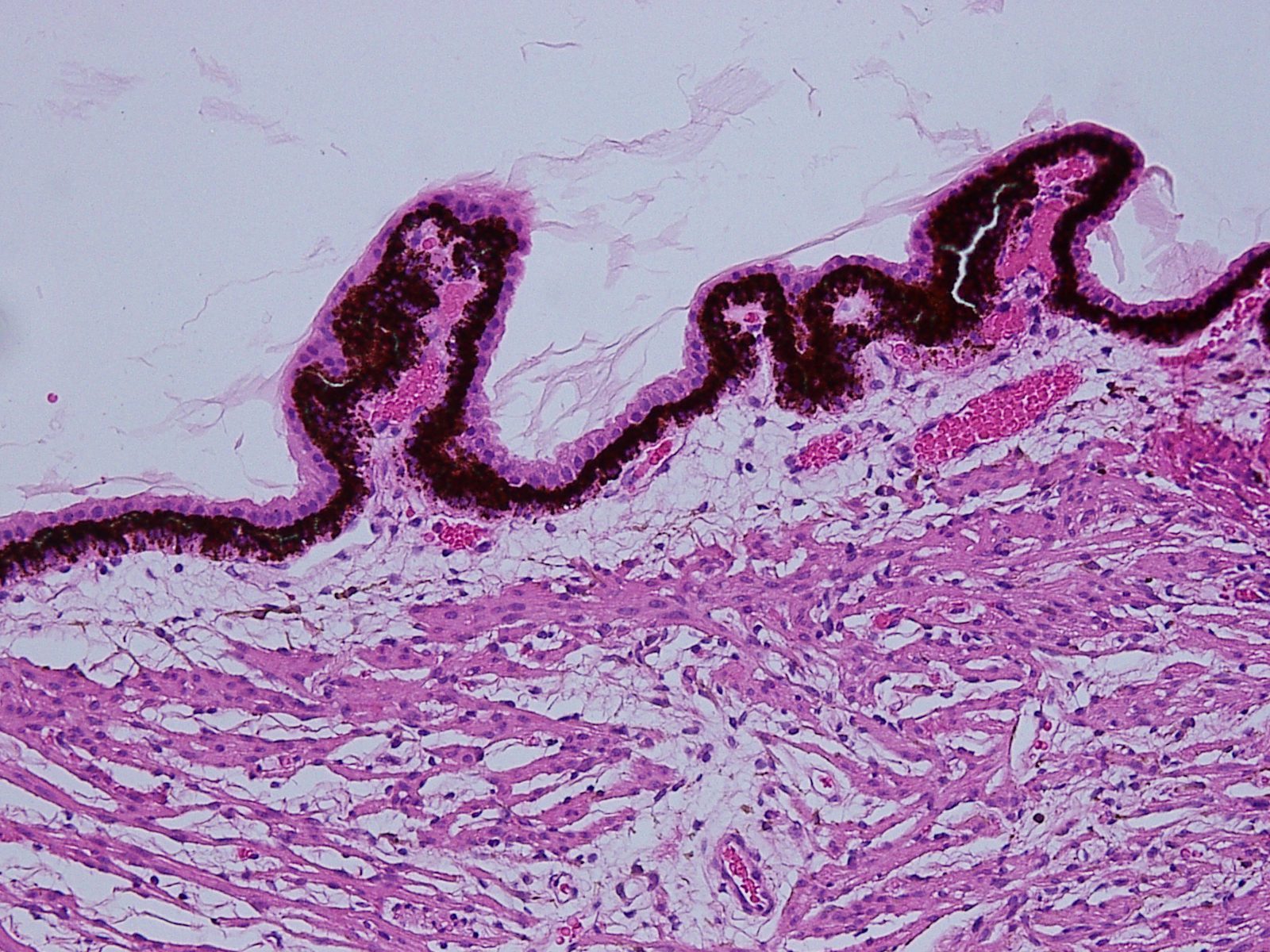
Ciliary Body

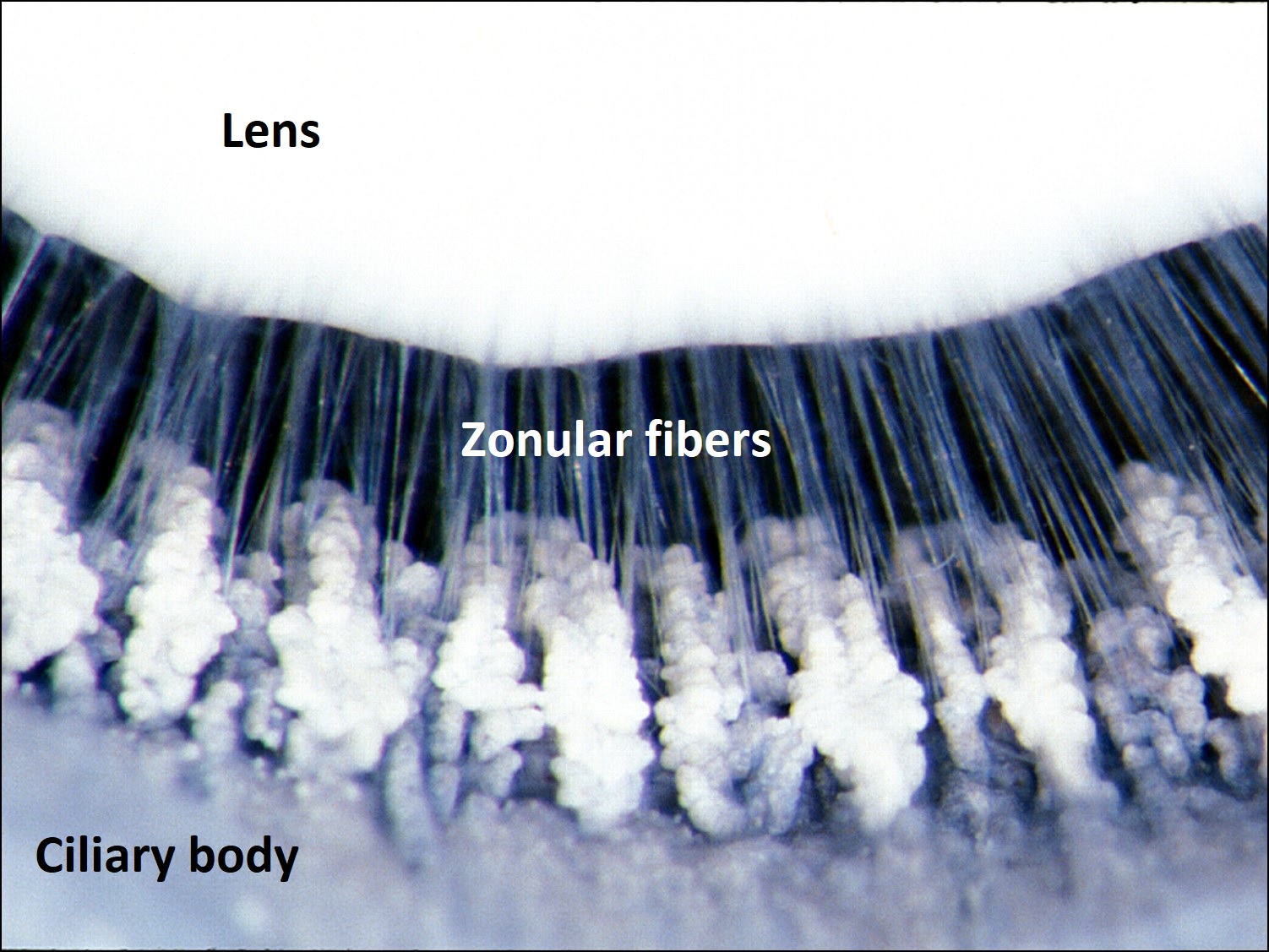
Photograph of ciliary body and zonules
Pars plicata

Pars plana


Transition from pars plana to retina and choroid at the ora serrata
Ciliary Body: 6-6.5mm wide–becomes continuous with choroid atora serrata
The ciliary body can be divided into 2 main areas:
- Pars plicata which contains the ciliary processesand zonular attachments–fimbriated
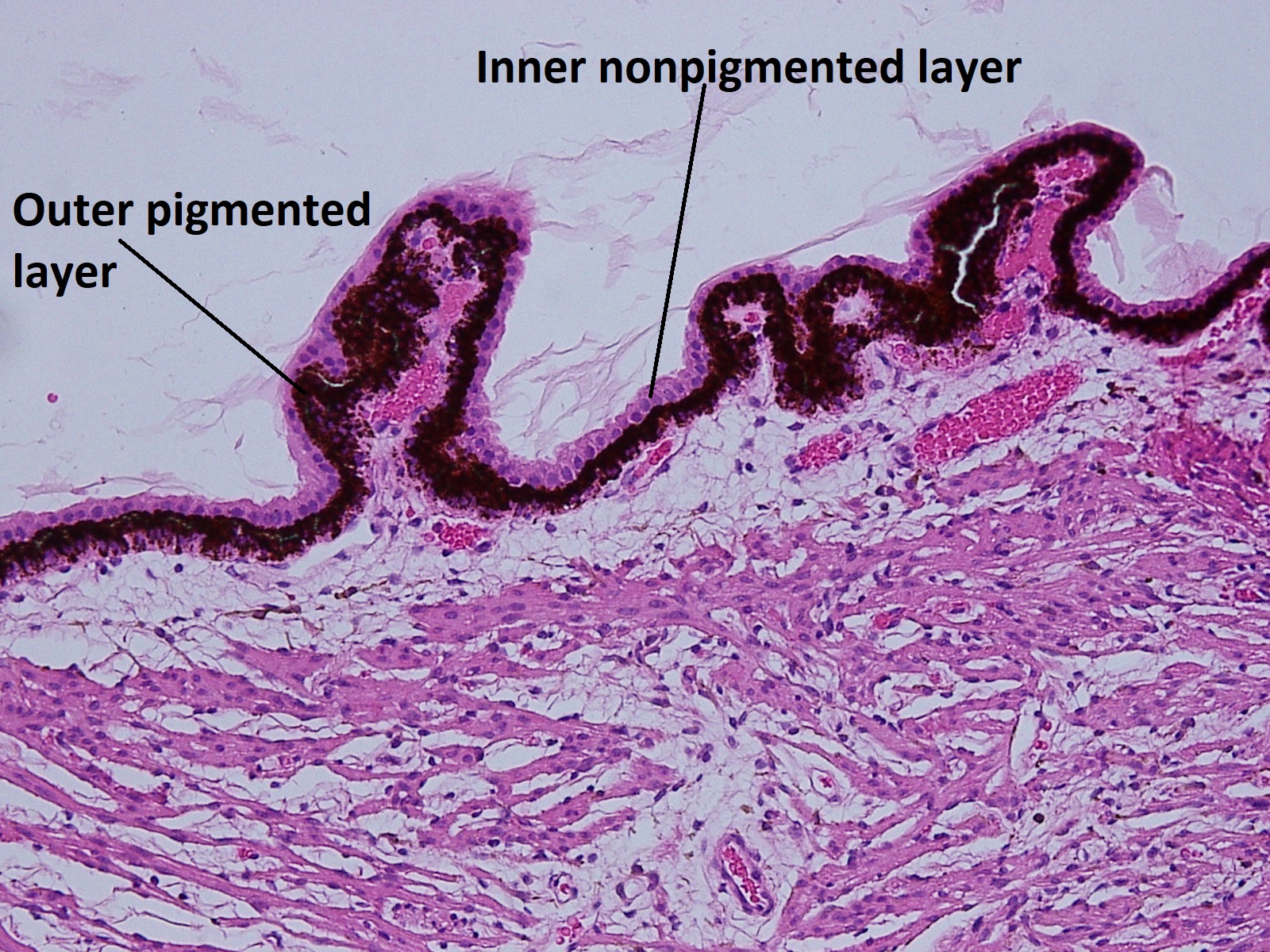
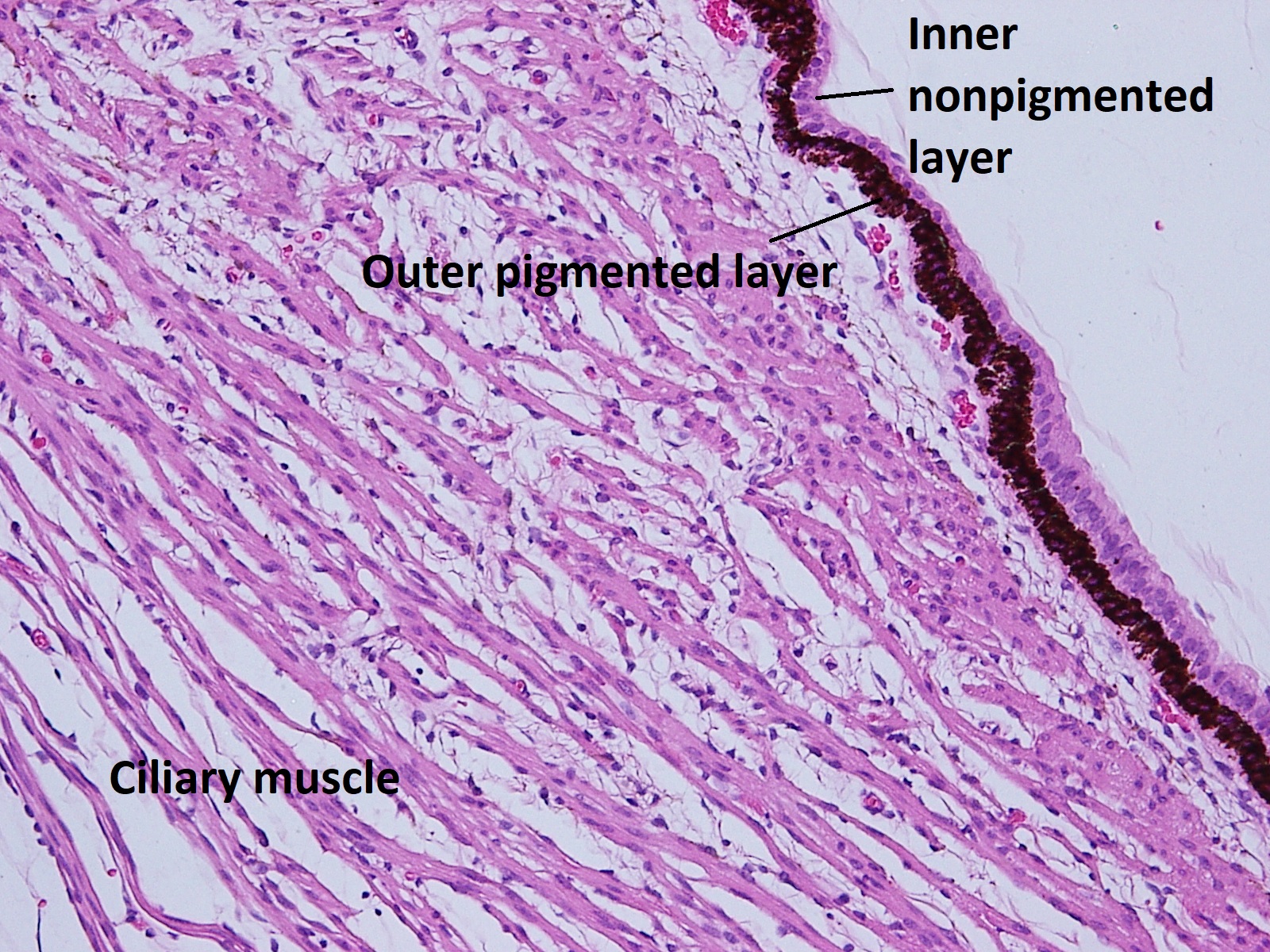
- Pars plana –smooth
- a. Outer pigmented layer
- b. Inner non-pigmented layer
Choroid


Normal choroid


Normal Choroid at the ora serrata
- Pigmented, vascular tissue that forms the middle coat of the posterior globe
- Extends from optic nerve to ora serrata
- 3 layers:
- Lamina fusca (suprachoroid layer)
- Stroma
- Choriocapillaris – provides nutrients for RPE + outer retina
Congenital Anomalies
Aniridia:
- True total absence of the iris is very rare
- More common to have an incomplete iris or narrow rim of iris tissue
- Usually bilateral
- Most aniridia due to PAX6 gene mutations
- About 85% have autosomal dominant familial aniridia –isolated to eye only
- Anterior chamber is often underdeveloped and aniridia is associated w/
- Cataract
- Corneal pannus
- Foveal hypoplasia
- Peripheral anterior synechiae are often present
- Glaucoma common
- AD + AR inheritance patterns exist
- Increased risk of Wilms tumor w/ 11p13 deletions + PAX6 mutations
- Also associated with microcephaly, mental retardation, genitourinary abnormalities
Nick’s Tips: Aniridia is associated with Wilms tumor. Look for this tumor of the renal system in patients with sporadic (non-familial) aniridia. The PAX6 gene and Wilms tumor gene (WT1) are closely associated on the short arm of chromosome 11.
Coloboma
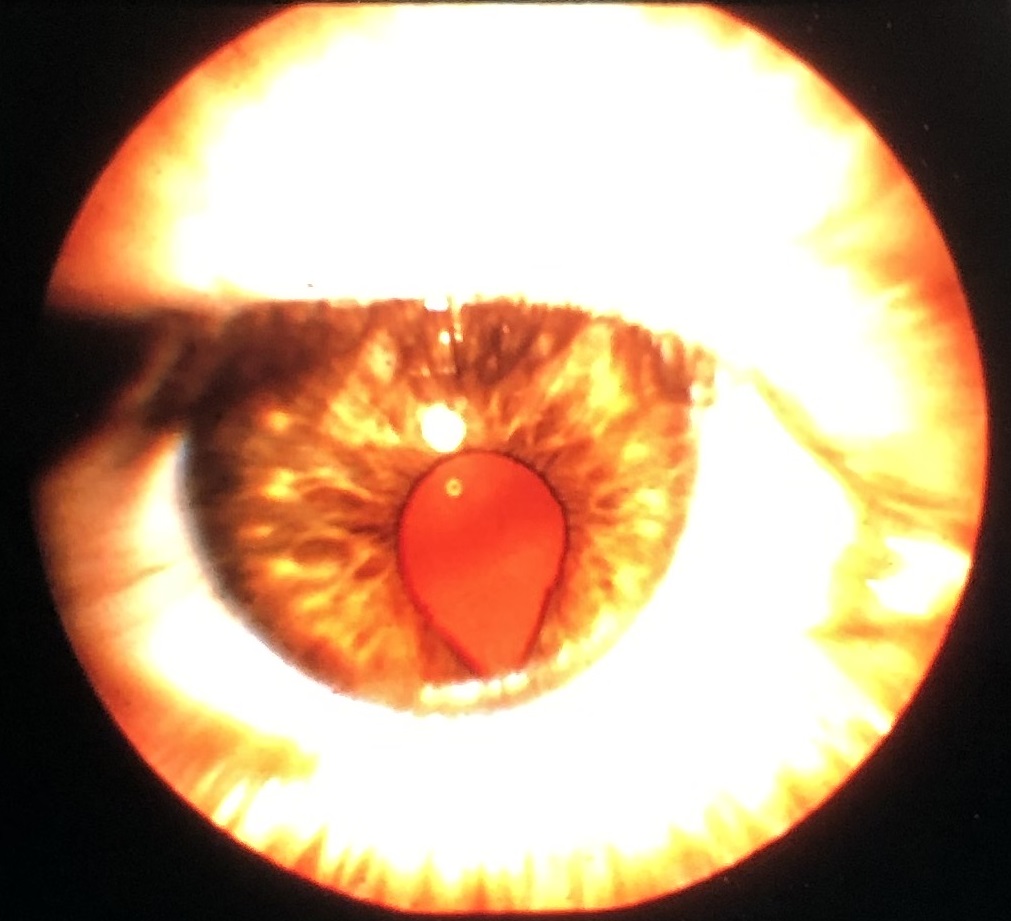
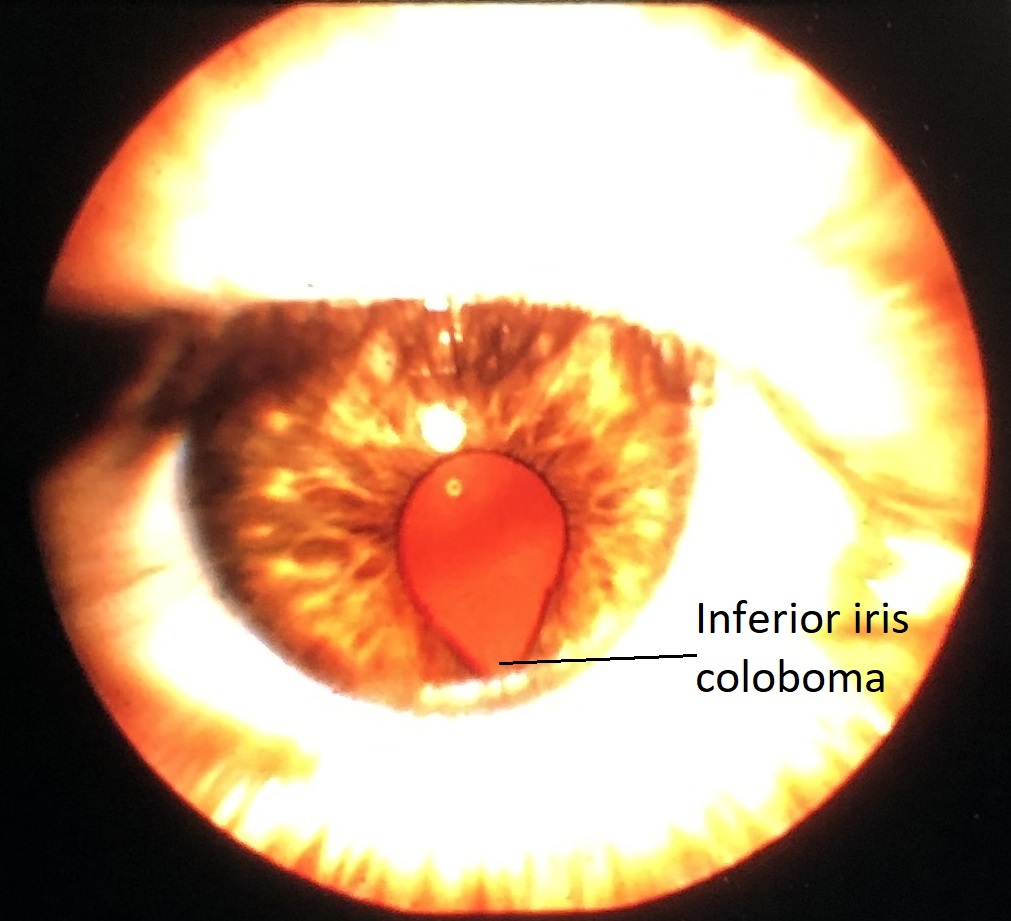
External photo of iris coloboma


Low magnification of iris coloboma histology
Coloboma: may affect iris, ciliary body, choroid or all 3
- May be isolated or part of larger syndrome
- Faulty closure of fetal fissure/optic groove
- Usually inferonasal
- 50% bilateral
Fuchs’ Adenoma


Fuchs’ Adenoma
- Acquired tumor of nonpigmented epithelium of ciliary body
- Possible association with sectoral cataract
- May stimulate other neoplasms of the iris and ciliary body
- Histopathology: sheets and tubules of hyperplastic, nonpigmented ciliary epithelium alternating with PAS-positive basement membrane material
Medulloepithelioma

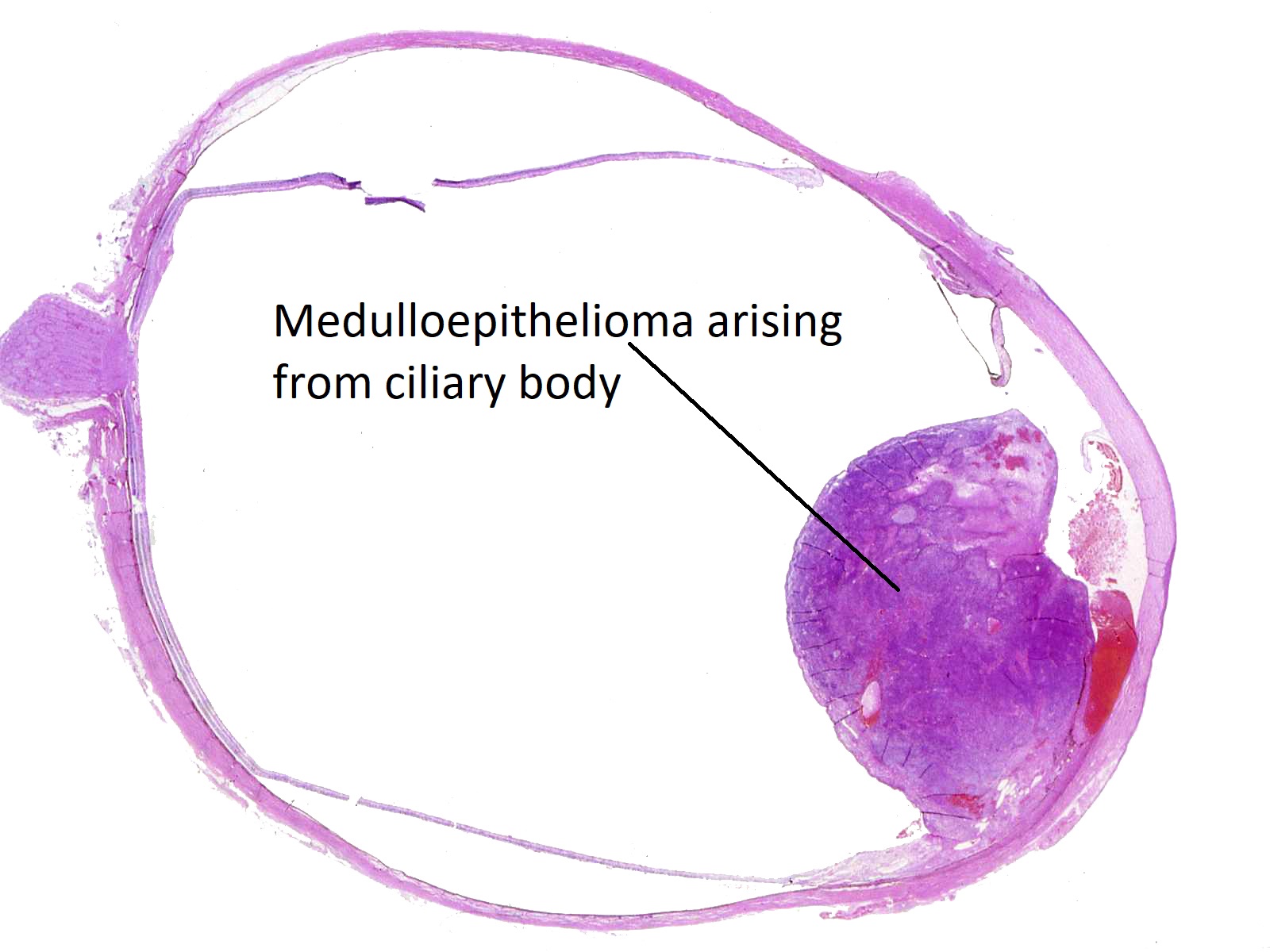
Ciliary body medulloepithelioma
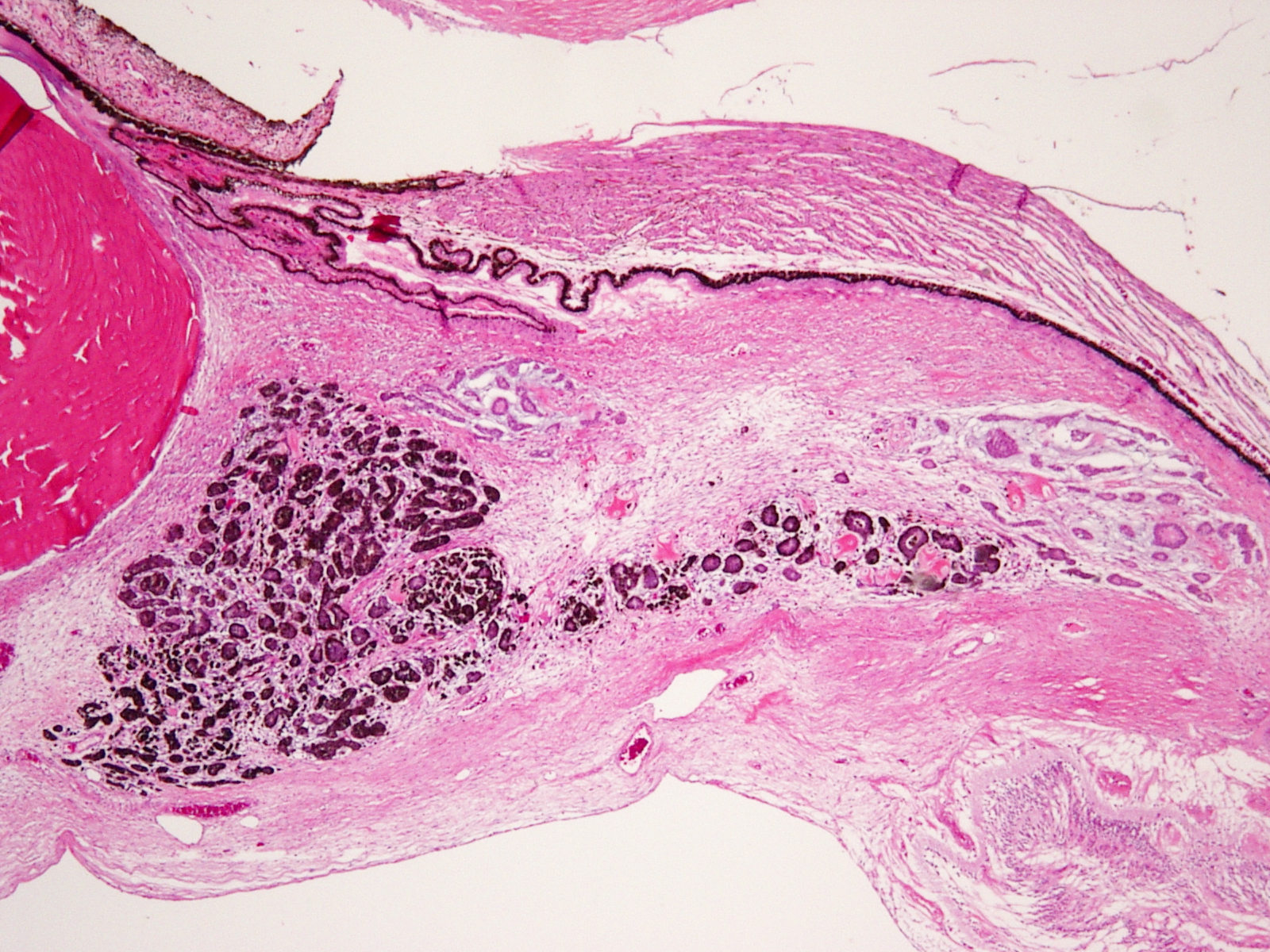
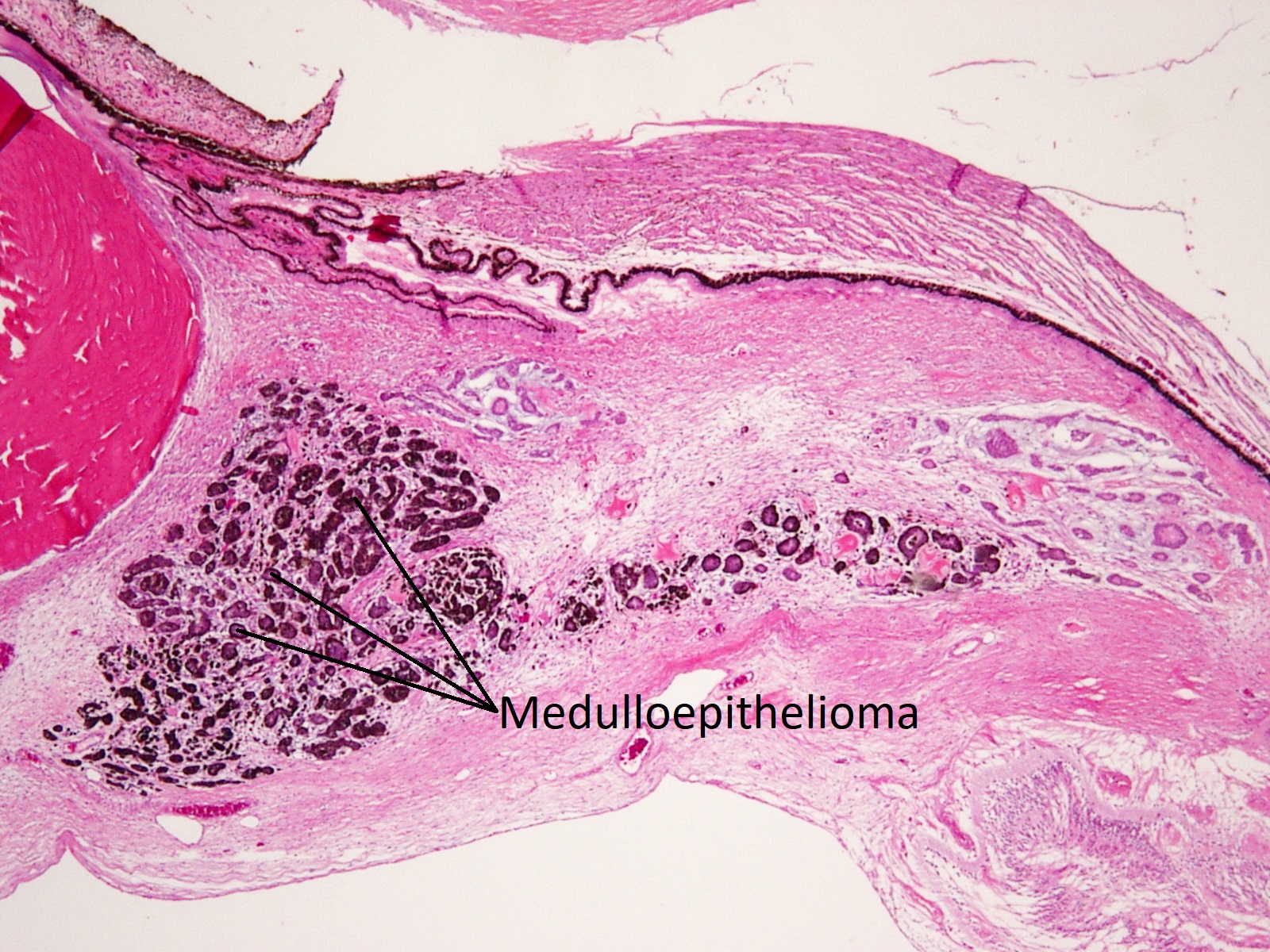
Ciliary body medulloepithelioma


Cords in medulloepithelioma

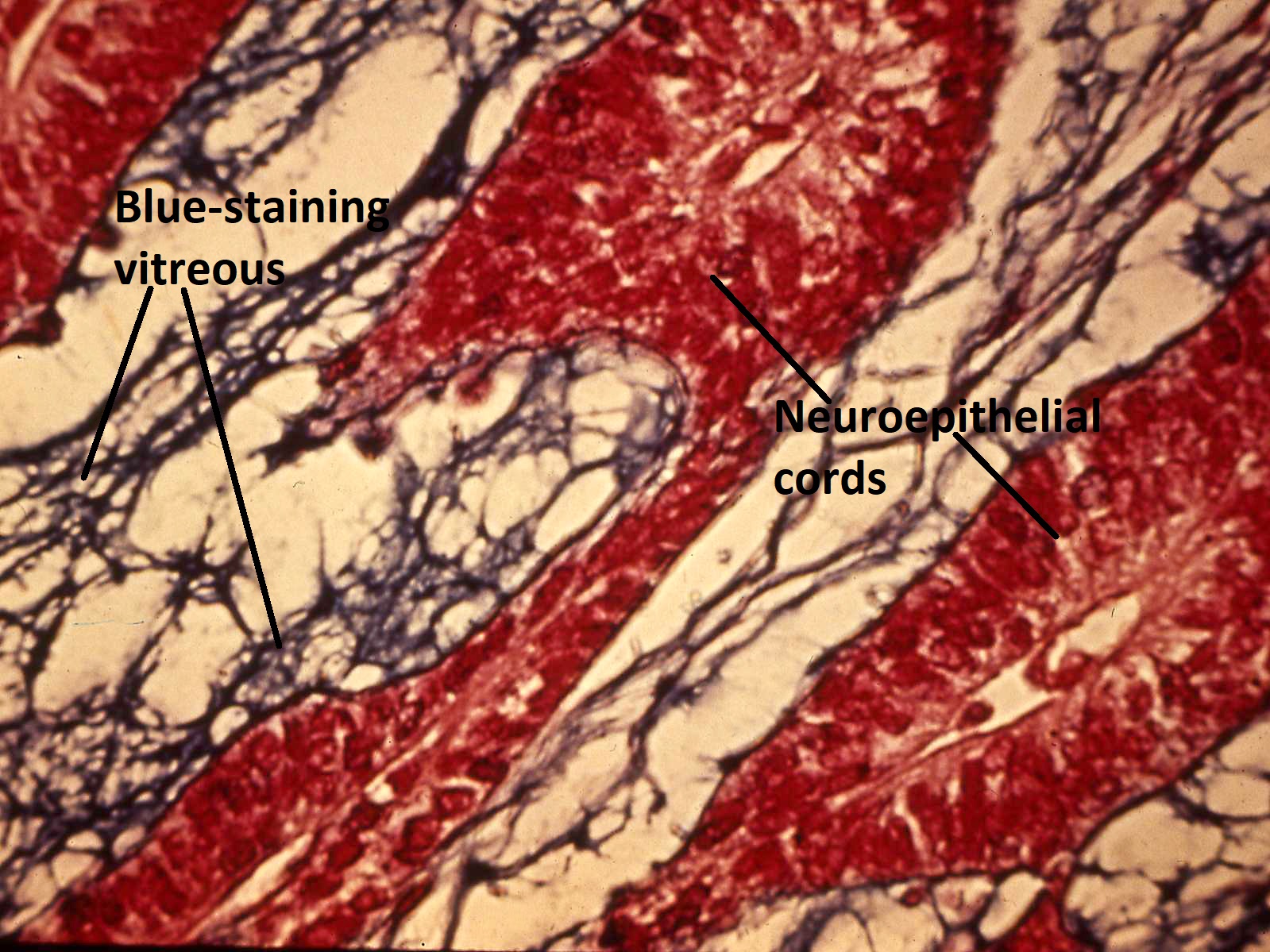
Alcian blue stain of medulloepithelioma


Medulloepithelioma

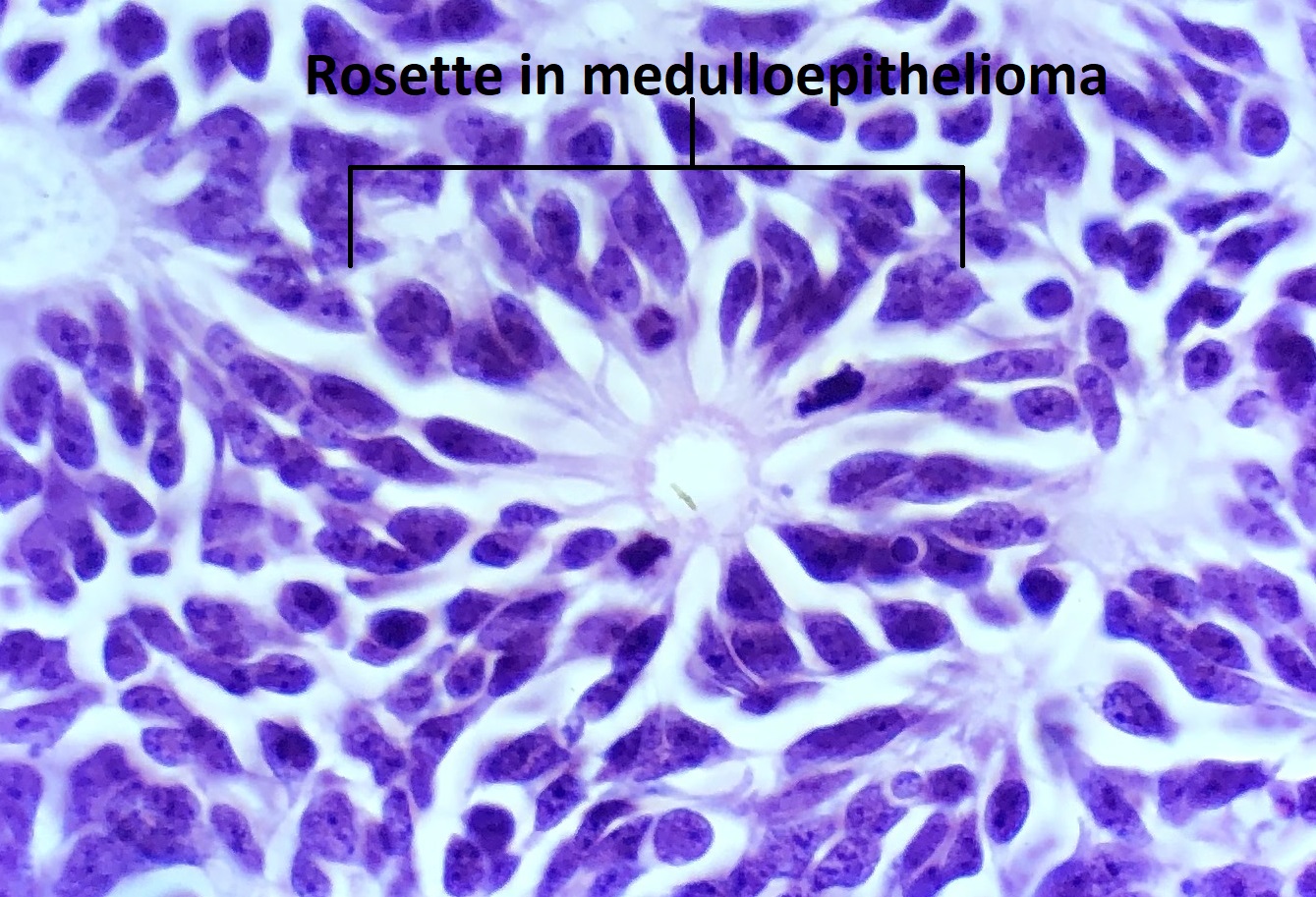
High magnification view of medulloepithelioma rosette


Masson’s trichrome stain of a teratoid medulloepithelioma
- AKA diktyoma
- Congenital neuroepithelial tumor from primitive medullary epithelium
- Usually in ciliary body, but rarely found in retina and optic nerve
- Clinical appearance: lightly pigmented to amelanotic cystic ciliary body mass eroding into anterior chamber and iris root
- Histology: ribbon-like structures of undifferentiated cells with little cytoplasm and distinct polarity
- 3-5 cell layers thick
- Basal surface lined with thin basement membrane
- Apical surface secretes mucinous hyaluronic acid-rich substance similar to vitreous
- Stratified sheets of cells form mucinous cysts
- Homer Wright and Flexner-Wintersteiner rosettes may be present
- Solid masses with neuroblastic cells difficult to distinguish from retinoblastoma
- Considered malignant if high mitotic rates and tissue invasion
- Enucleated patients have high survival rates
- Typically follows relatively benign course if tumor remains in eye
- Teratoid medulloepitheliomas: tumor with cells from 2 different embryonic germ layers
- Malignant teratoid medulloepitheliomas have solid areas of neuroblastic cells and sarcomatous heteroplastic elements (i.e. cartilage, smooth muscle)
Inflammation of the Choroid
May be infectious or non-infectious –for moreinformation, please review section on uveitis.
Infectious:
- Generalized or localized
- Exogenous –from outside
- Endogenous –hematogeneous seeding
Non-infectious:
Sympathetic Ophthalmia


Diffuse choroidal inflammation in sympathetic ophthalmia


Histology showing choroidal inflammation
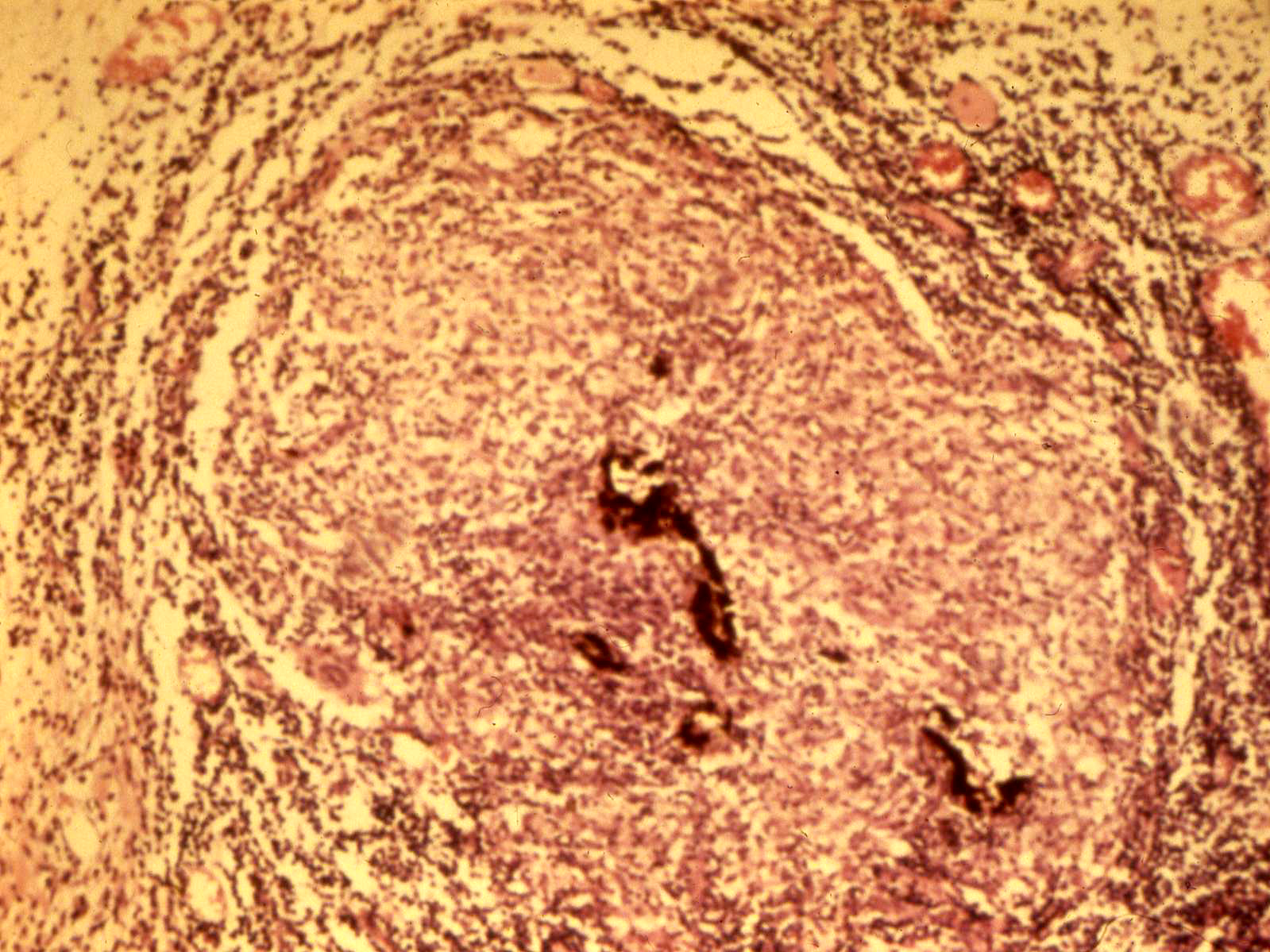

Granuloma formation in sympathetic ophthalmia

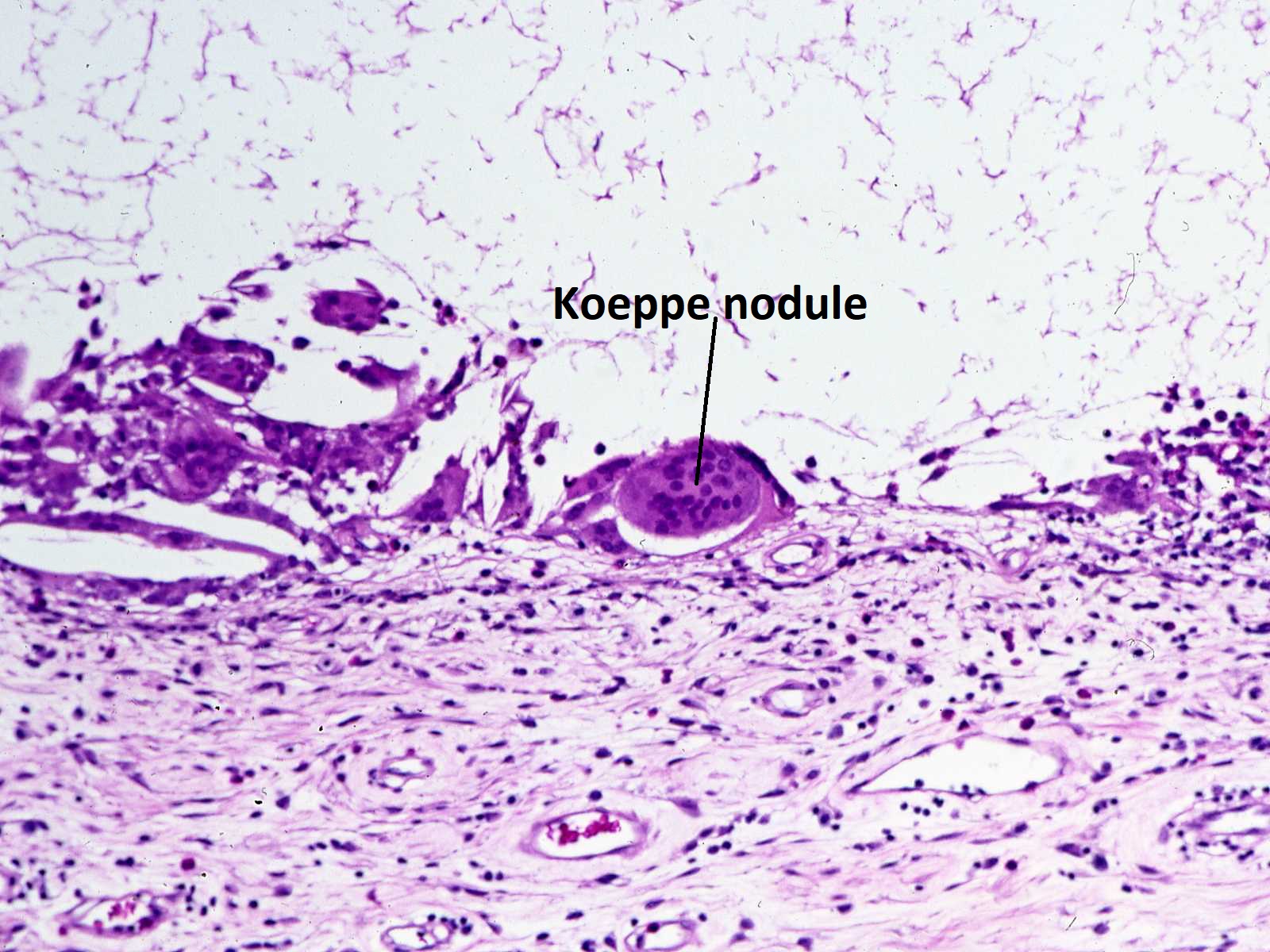
Histology of Koeppe nodule

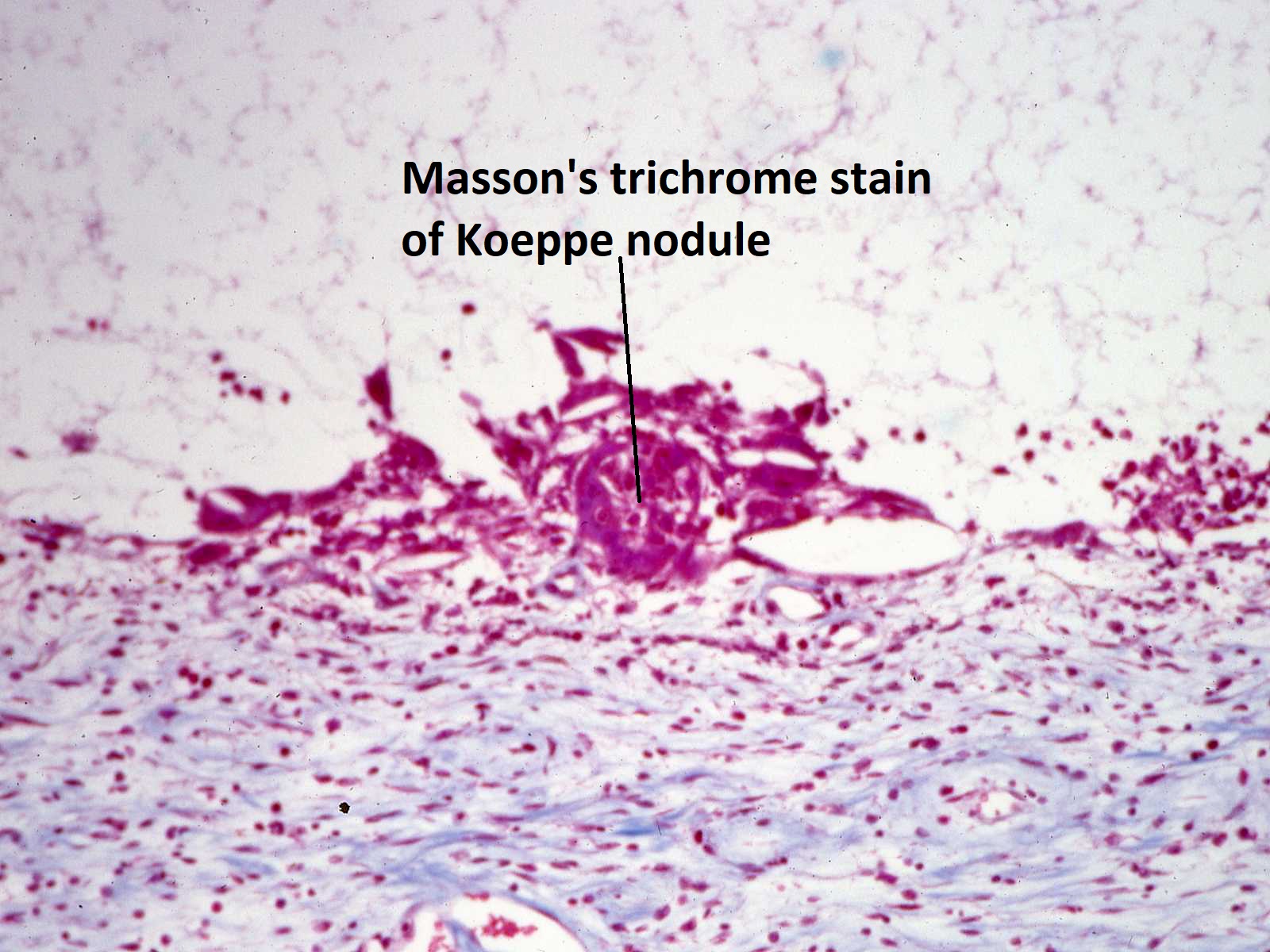
Masson’s trichrome stain of Koeppe nodule


High magnification view of a Masson’s trichrome stained Koeppe nodule
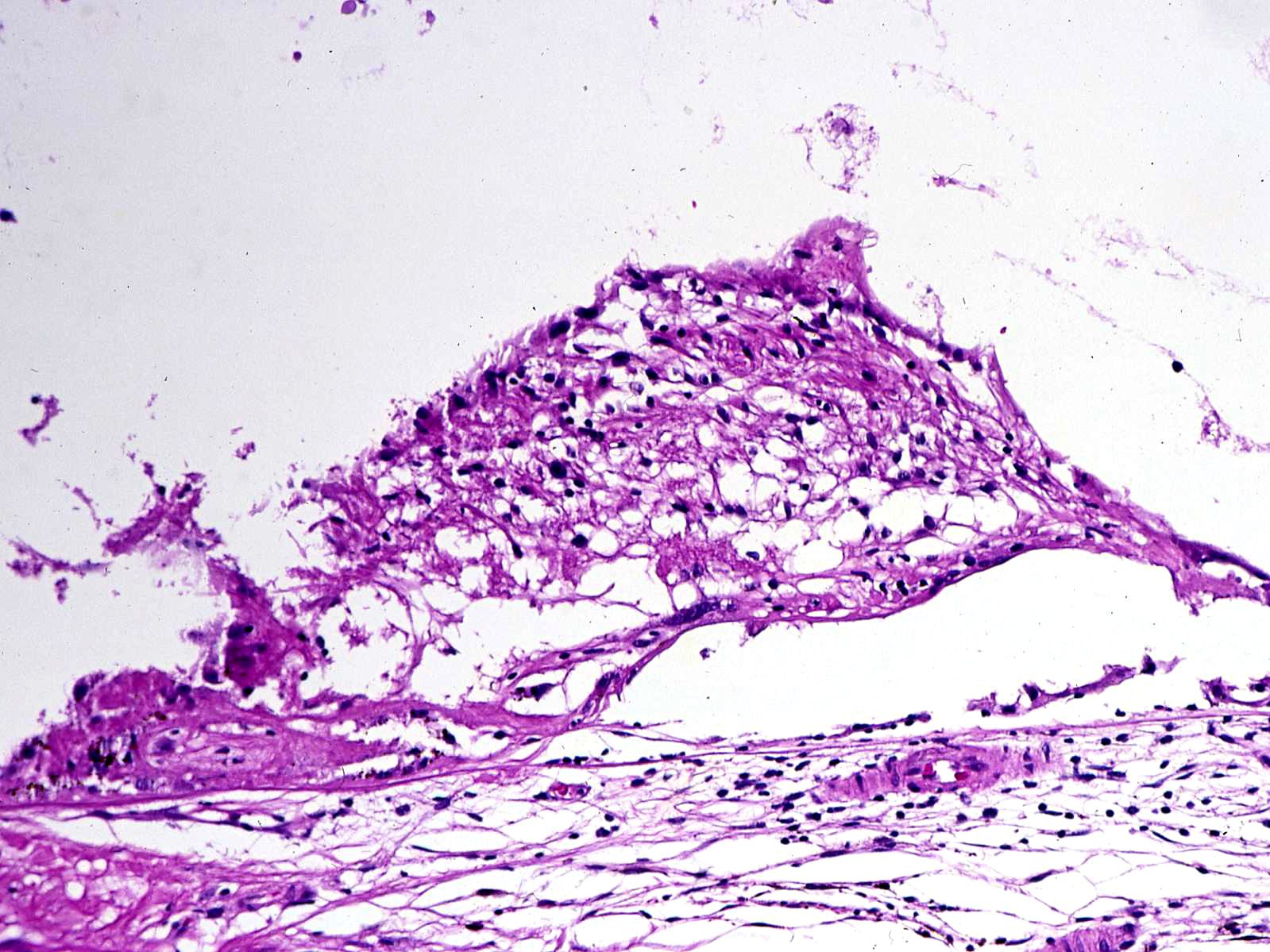

Histology of Dalen-Fuchs nodule
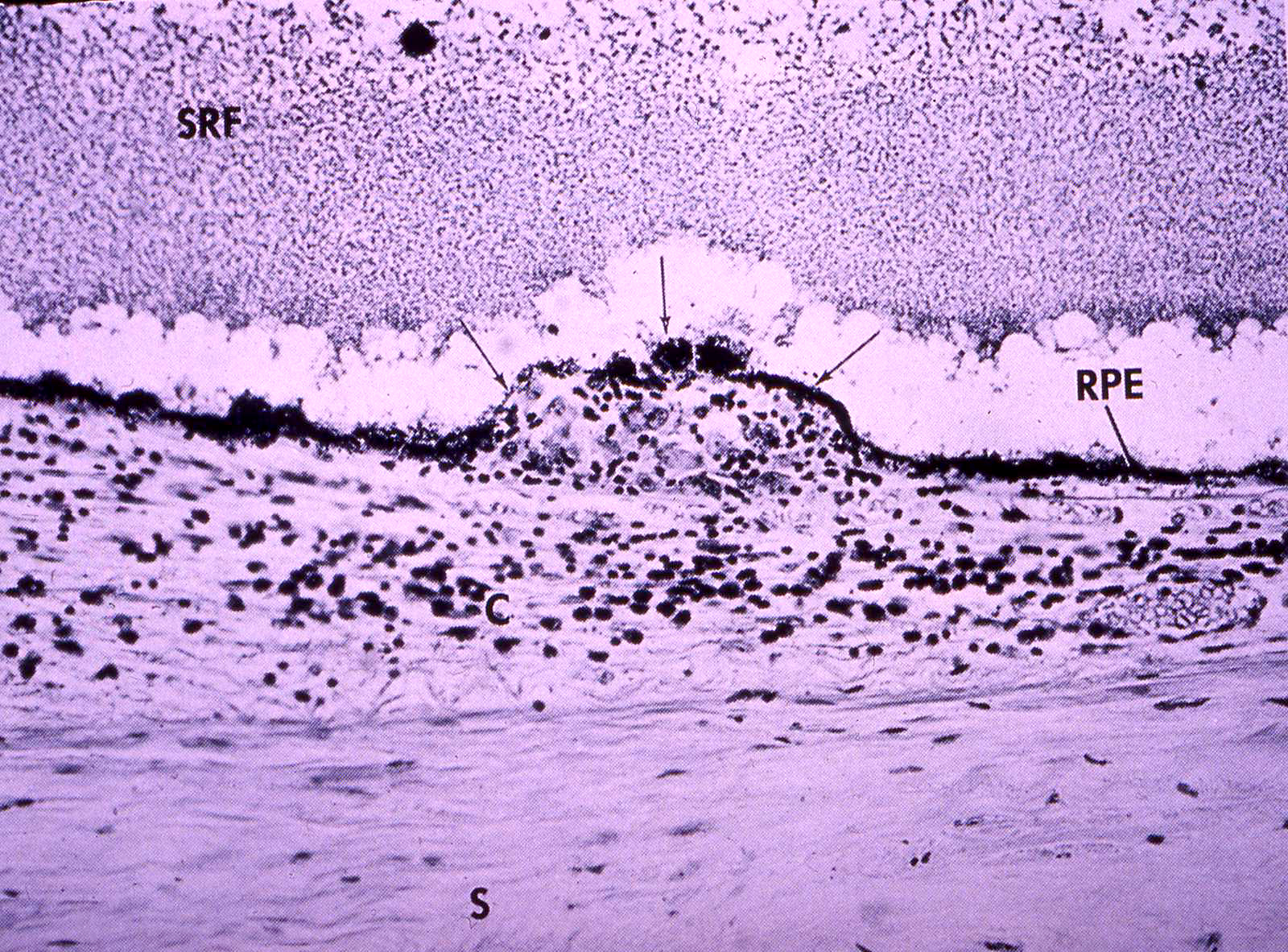

Anatomic location of Dalen-Fuchs nodule
- Bilateral panuveitis, occurring after severe injury to one eye
- May occur days or up to 50 years later (typically 4-12 weeks latency, 90% within 1 year)
- Histopathology shows diffuse thickening and granulomatous reaction of uveal tract, typically sparing choriocapillaris and retina
- T-cell Lymphocytes + epitheliod histiocytes w/phagocytosed melanin
- May have mutton fat KPs + A/C inflammation
- Koeppe nodules-inflammatory cell precipitates at the pupillary margin
- Dalen-Fuchs nodules-epithelioid cells localized between RPE + Bruch’s (also occur in sarcoidosis and VKH)
- The cause is unknown. May represent hypersensitivity to melanin pigment, retinal S –protein, or other retinal proteins. Injury allows immunologic access to privileged uveal proteins leading to immunologic destruction in eye.
- Removal of severely injured eye within 1 week will usually prevent development of sympathetic ophthalmia.
Nick’s Tips: look for granulomatous inflammation with history of old injury or surgery to same or contralateral eye. Look for epithelioid histiocytes and giant cells scattered in plasma cells and lymphocytes.
Vogt –Koyanagi –Harada Syndrome
- Posterior or panuveitis, +/-systemic symptoms
- Most common in Asian or Native American
- Usually age 30-50
- Bilateral decreased visual acuity, pain, redness, photophobia
- Systemic -> poliosis (loss of pigment in lashes), vitiligo, dysacusis, HA, seizure
- Chronic, diffuse, granulomatous uveitis -> Exacerbation + Remission
- No sparing of choriocapillaris
Sarcoidosis
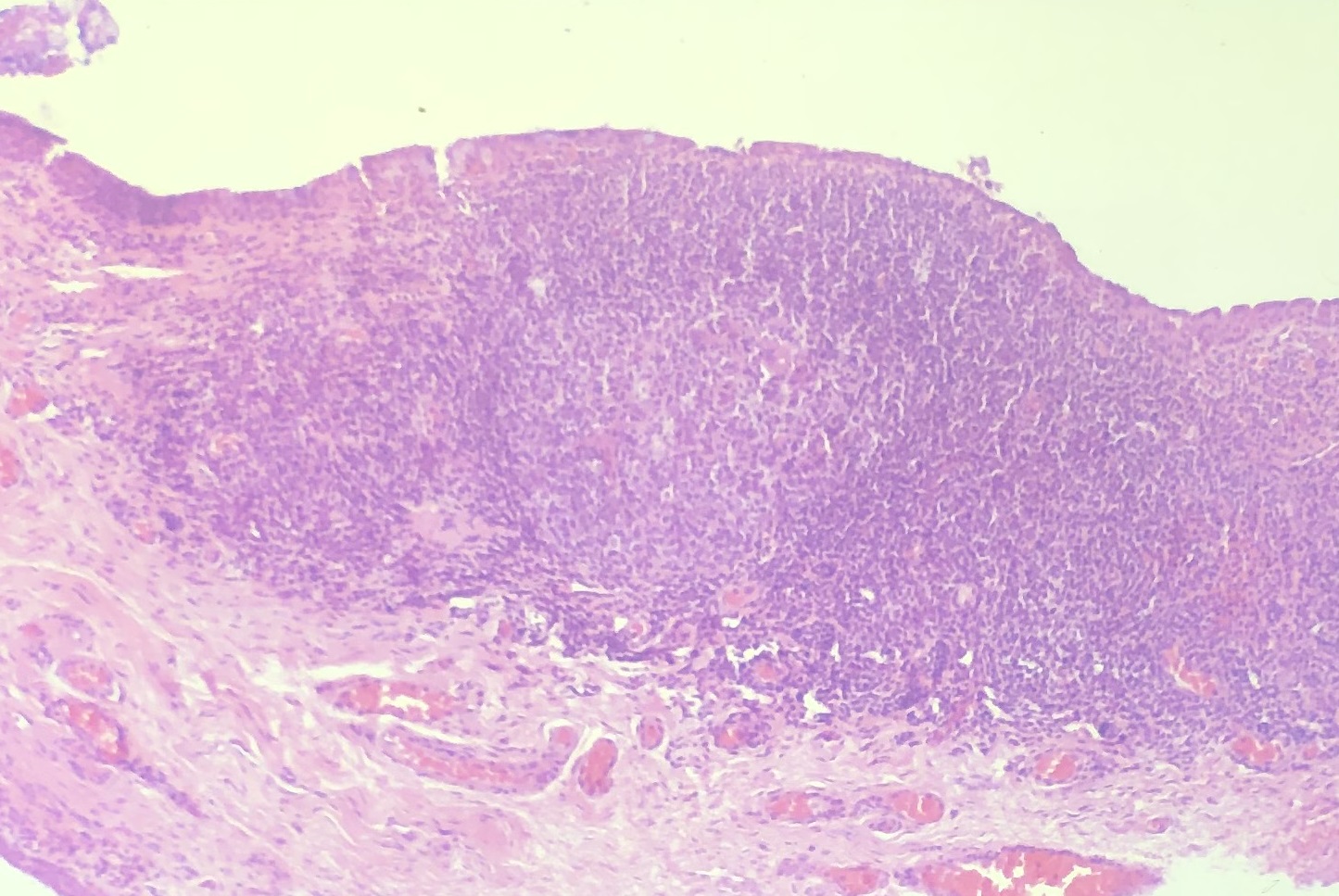

Noncaseating granuloma in sarcoidosis

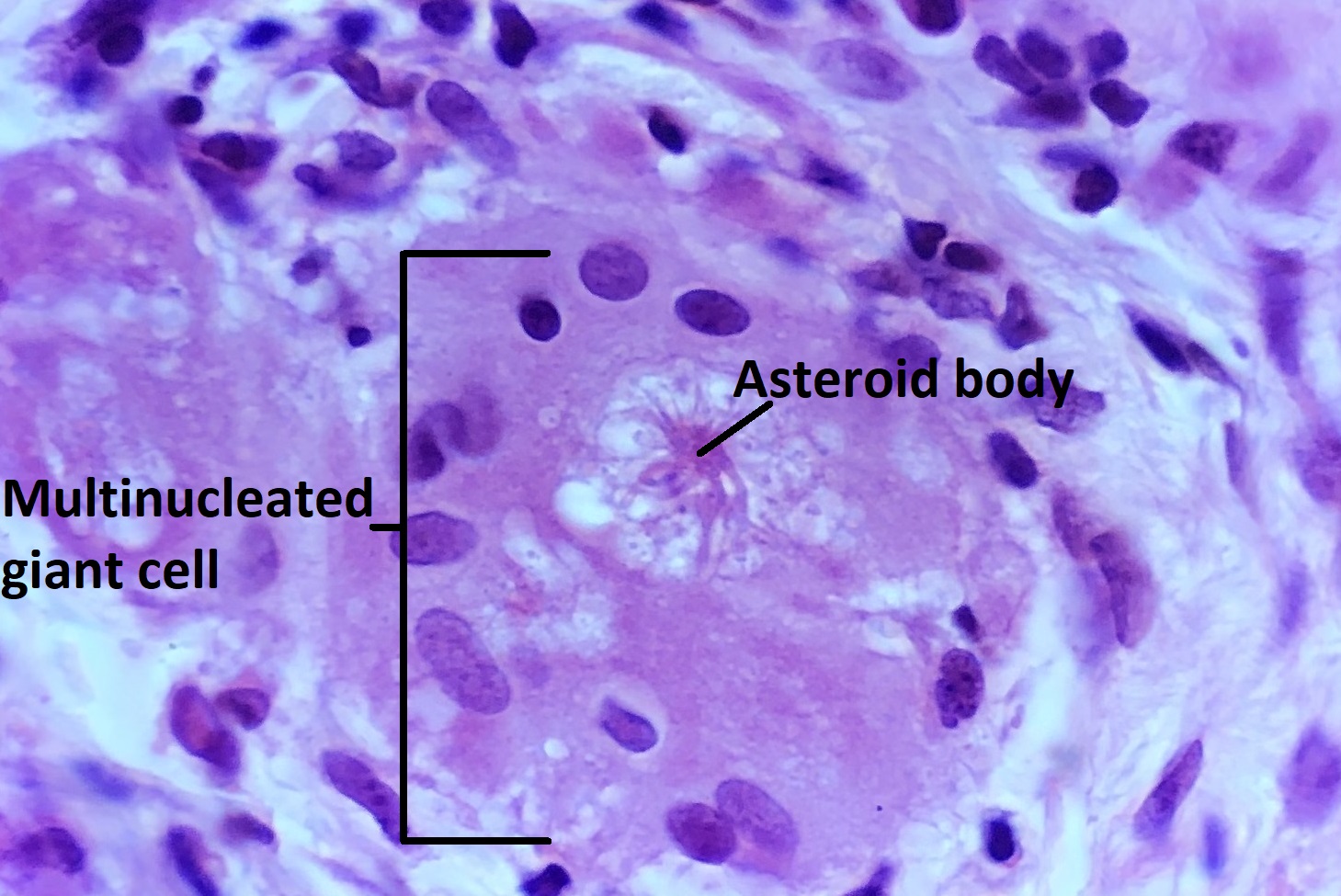
Asteroid body in a multinucleated giant cell
- Multisystem disease w/ ocular inflammation
- Noncaseating histiocytes + lymphocytes = granulomatous inflammation
- Anteriorly
- Koeppe nodules at pupil margin
- Busacca nodules found elsewhere on iris
- Posteriorly
- Chorioretinitis
- Phlebitis-manifest as “candlewax lesions”
- Optic nerve edema secondary to inflammation
- Giant cells
- Asteroid bodies (star-shaped,acidophilic)
- Schaumann bodies (spheroid, basophilic)
Nick’s Tips: Clinically, granulomatous inflammation is associated with “mutton fat” keratic precipitates. Histologically, we see aggregated epithelioid histiocytes and/or inflammatory giant cells. Granulomatous inflammation is caused by certain diseases, so must search for sarcoidosis, acid-fast organisms like TB, and spirochetes. Sarcoidosis is a common cause of granulomatous inflammation –look for discrete, well circumscribed, histiocytes with giant cells separated from lymphocytes with sharp demarcations.
Rubeosis iridis

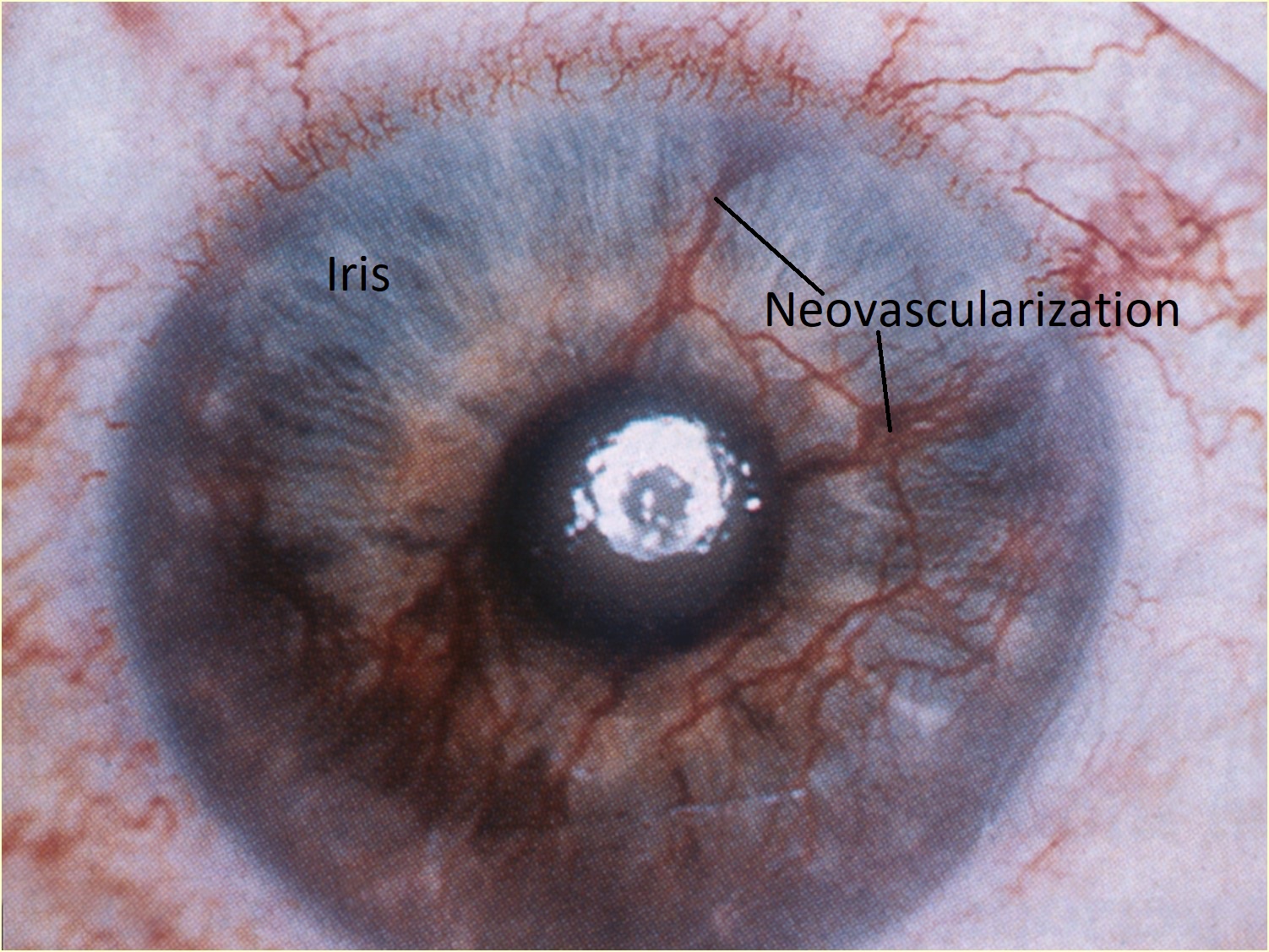
Rubeosis iridis
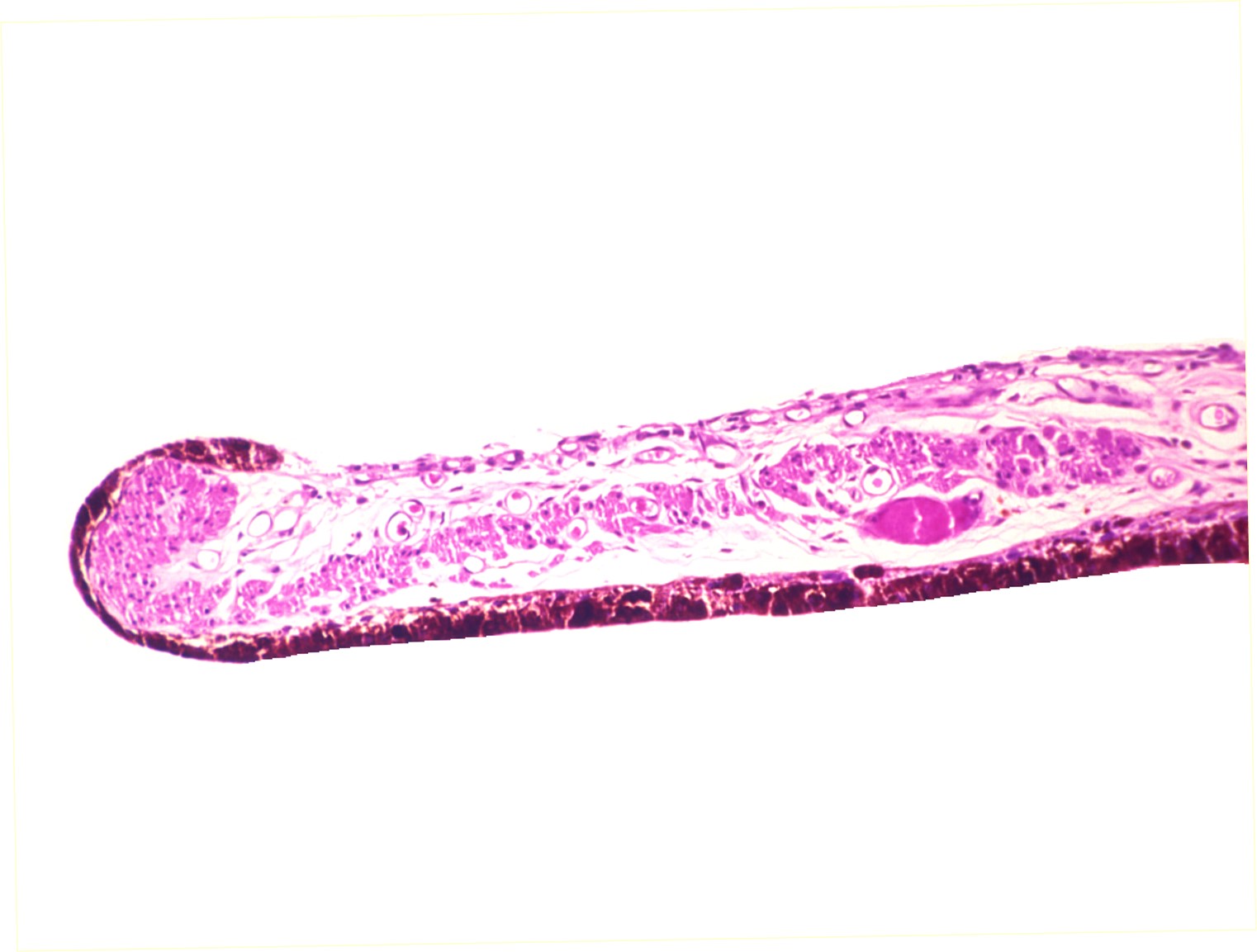

Histology of iris neovascularization


Neovascularization at the angle
Rubeosis iridis (NVI) can cause neovascular glaucoma
- Usually vessels arise on anterior surface of the iris but may also arise on posterior surface
- Fibrovascular tissue surrounds vessels and may lead to ectropion uvea (displacement of iris to level of sphincter) or PAS
- Advanced stages: dilator atrophy, stromal fibrosis, attenuation of PEN on progressive rubeosis causes small tufts at the iris margin
Rubeosis is associated with
- Diabetes
- CRVO
- BRVO
- CRAO
- Carotid occlusive disease
- Sickle cell disease
- Intraocular inflammation
- Retinal detachment
- Retinal detachment surgery
- Coats Disease
- Retinoblastoma
- Melanoma
- Radiation
- Trauma
- Secondary glaucoma
Hyalinization of Ciliary Body
- Process of normal aging
- Thin processes of pars plicata become blunted and undergoeos inophilic changes
- Contributes functionally to development of presbyopia
Choroidal Neovascularization (CNV)


Sub-RPE and subretinal neovascularization


Histopathology of sub-RPE neovascularization
Photo reference: Harper RA. Basic Ophthalmology. Amer Academy of Ophthalmology; 2010.
CNV may be caused by any condition that disrupts the Bruch’s Membrane such as:
- Age-related macular degeneration
- Angioid Streaks
- Ocular Histoplasmosis
- Surgery / Laser
- Trauma
Complications include: disciform scarring, serous or hemorrhagic detachment
Polypoidal Choroidal Vasculopathy
- AKA Posterior Uveal Bleeding Syndrome and Multiple Recurrent Serosanguinous RPE Detachments
- Thin-walled, dilated vascular channels between RPE and outer layers of Bruch membrane
- Associated with subretinal hemorrhages, hemorrhagic RPE detachment, and serosanguinous RD
- Arise from short posterior ciliary arteries
- Choroidal neovascularization often present
- Hyperfluorescent polypoidal lesions present on FA without leakage
Iris Nevus
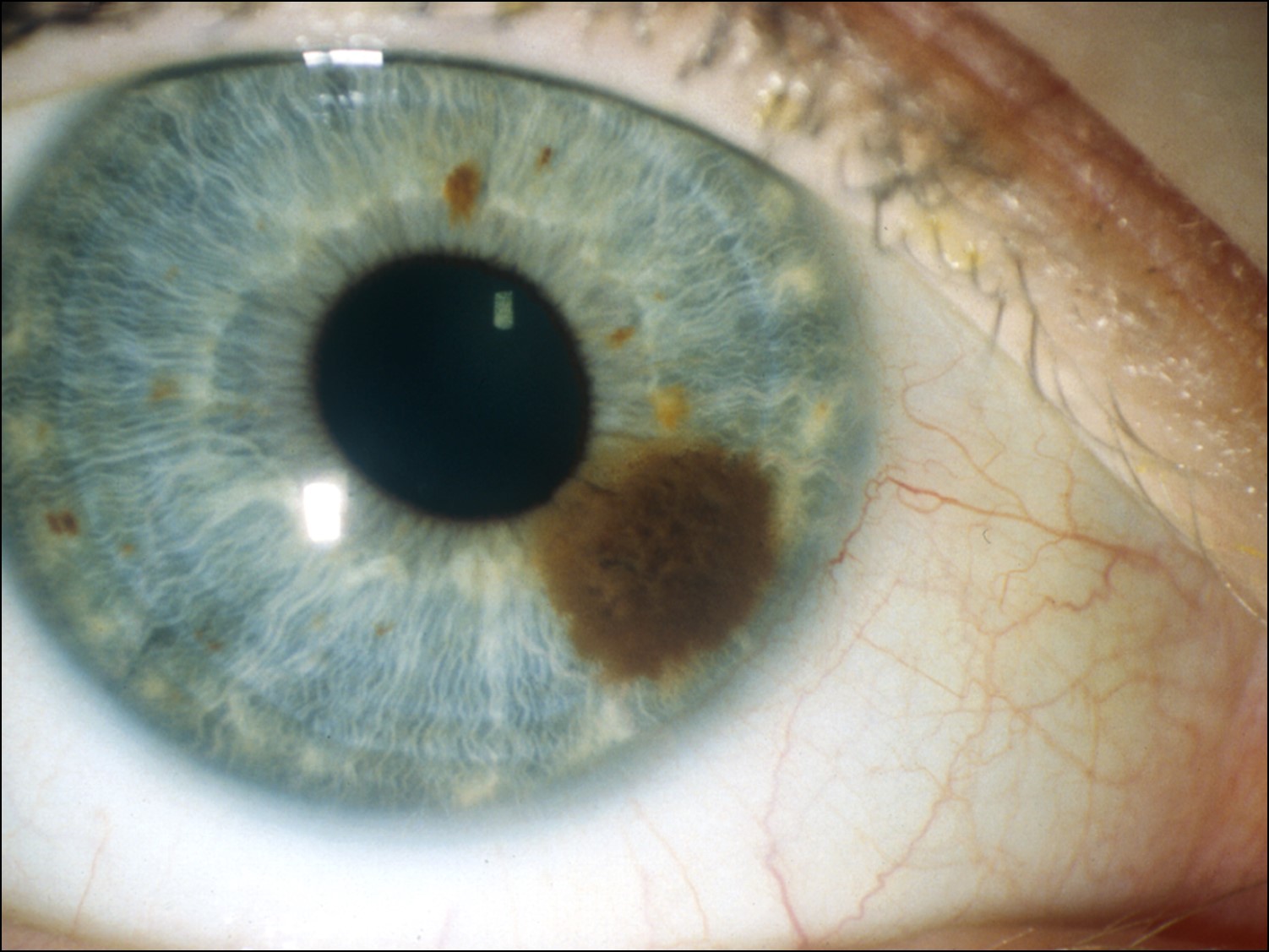

External photo of iris nevus

Histology of iris nevus


Histology of iris nevus

High magnification of iris nevus
Iris Nevus: increased incidence w/ neurofibromatosis type 1
Histologically leads to accumulations of dendritic or spindle cells
No cellular atypia ormitotic activity
No treatment – observation only
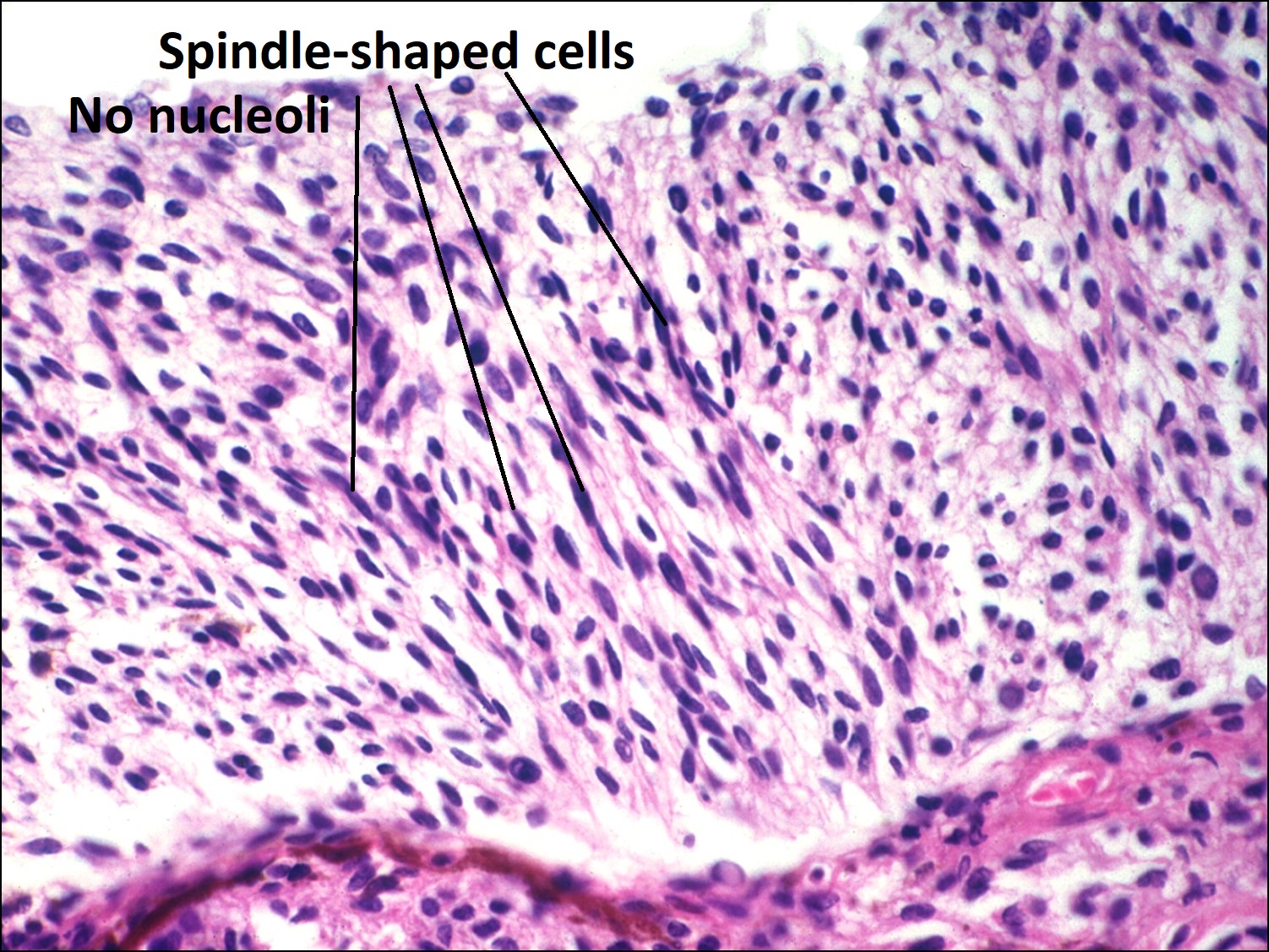
Iris Melanoma
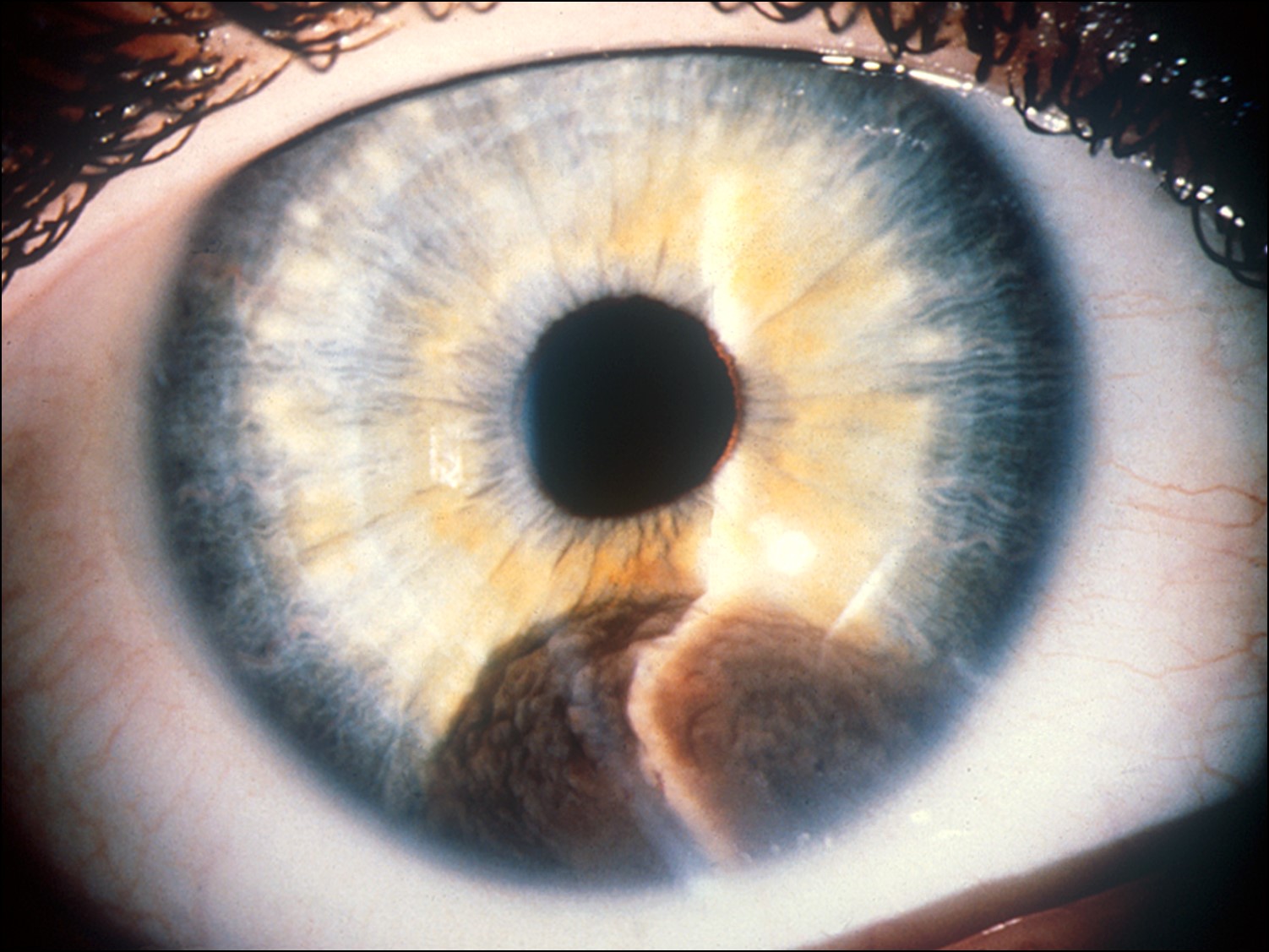

External photo of iris melanoma
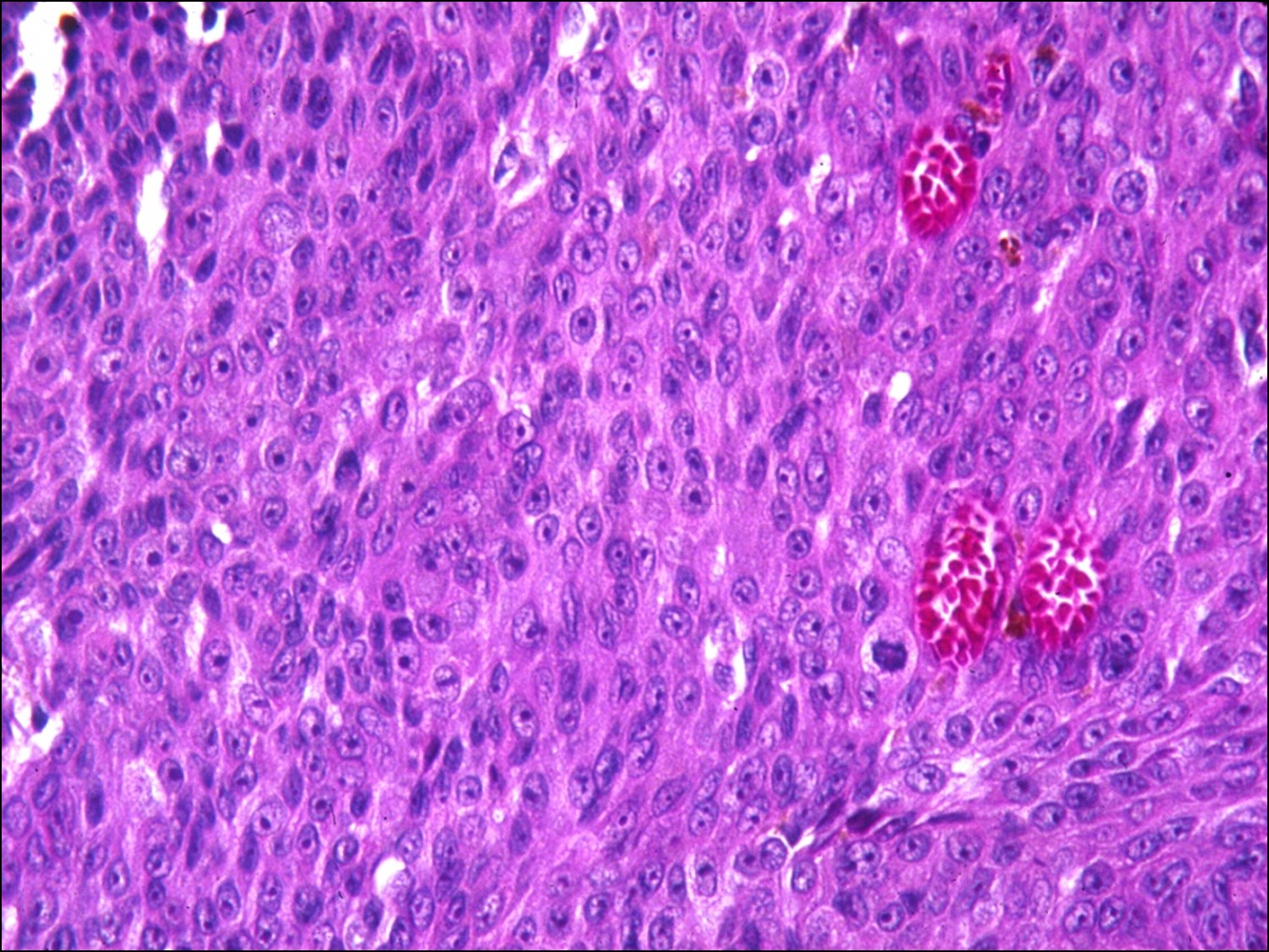

Histology of iris melanoma


Histology of iris melanoma


High magnification of iris melanoma
- Usually much less aggressive that posterior melanomas
- Usually in inferior sector
- Rarely leads to spontaneous hyphema
- Age 40-50 years (10-20 years younger than posterior melanomas)
- May be locally aggressive, but rarely metastasize
- May be part of a ring melanoma arising from the ciliary body –much worse prognosis
- Cataract and secondary glaucoma possible.
Choroid + Ciliary Body Neoplasms
Choroidal Nevus


Color photo of choroidal nevus
Nevus (usually located in the choroid, nevi are rare in ciliary body)
- 4 cell types:
- Plump polyhedral
- Slender spindle
- Plump fusiformdendritic
- Balloon cells
- May cause local mess effects
- Usually remain fixed over long observation
- Do have malignant potential
Nick’s Tips: very common –about 5% of patients. Risk of melanoma low (about 1 in 10,000 to 1 in 15,000) if no risk factors, but rises to about 38% with one risk factor and about 50% with 2 risk factors.
Risk factors for melanoma include the following:
- Orange pigment (lipofuscin)
- Serous retinal detachment
- Symptomatic
- Greater than 2mm thickness as measured with ultrasound
- Posterior margin of the nevus contacts the optic disc
Melanocytoma of the choroid


Low magnification of choroidal melanocytoma
Melanocytoma:
- Also known as magnocellular nevus (specific type of nevus)
- Jet Black, most common peripapillary
- Heavy pigment. May have to bleach sections to see architecture
- May be found in all racial backgrounds –up to 50% in African American in USA
- Typically arise from the optic nerve, but may arise anywhere in uveal tract
- May undergo spontaneous necrosis and can be recognized by the presence of melanin laden macrophages
- May sometimes cause secondary glaucoma due to necrosis of iris melanocytoma and subsequent blocking of the trabecular meshwork by pigment laden macrophages. Can be seen with gonioscopy.
- Transformation to melanoma is rare
Nick’s Tips: Very dark and heavily pigmented lesions often associated with the optic disc. You may need to bleach specimen in order to see details. When looking at bleached specimens –will see bland nuclei and a low nuclear to cytoplasmic ratio.
Choroidal Melanoma


Photo of choroidal melanoma
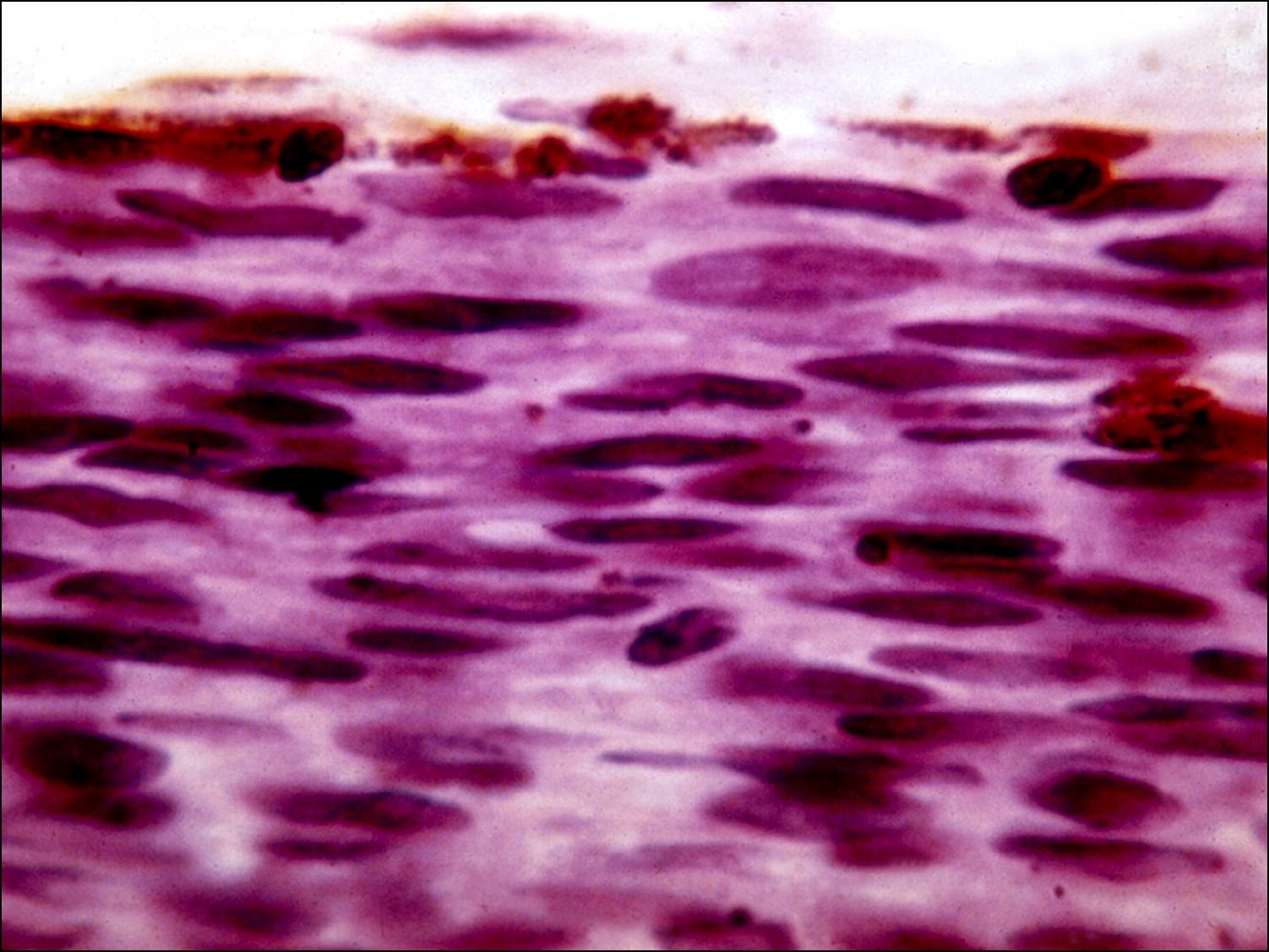
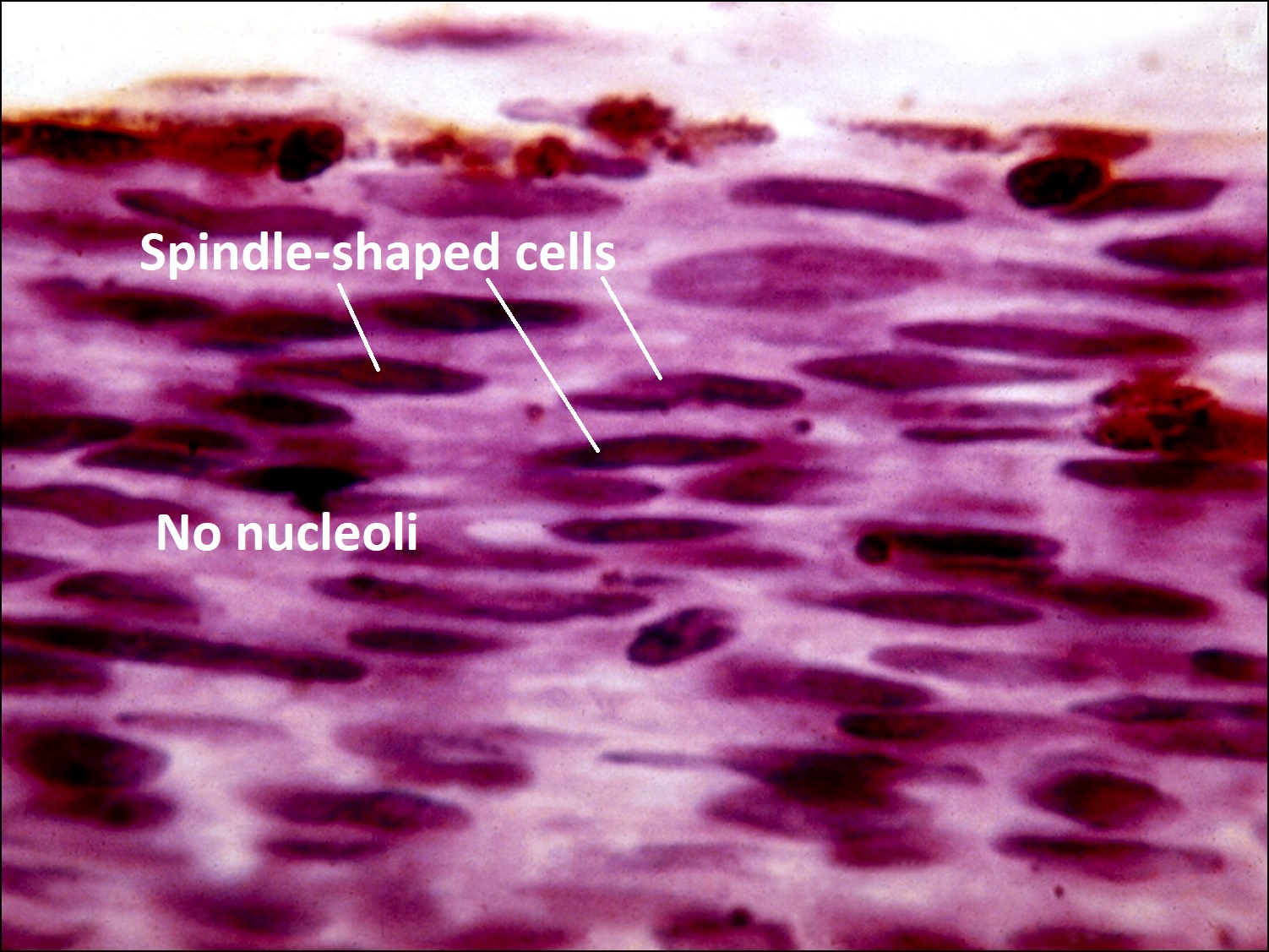
Histology of choroidal melanoma


Histology of choroidal melanoma
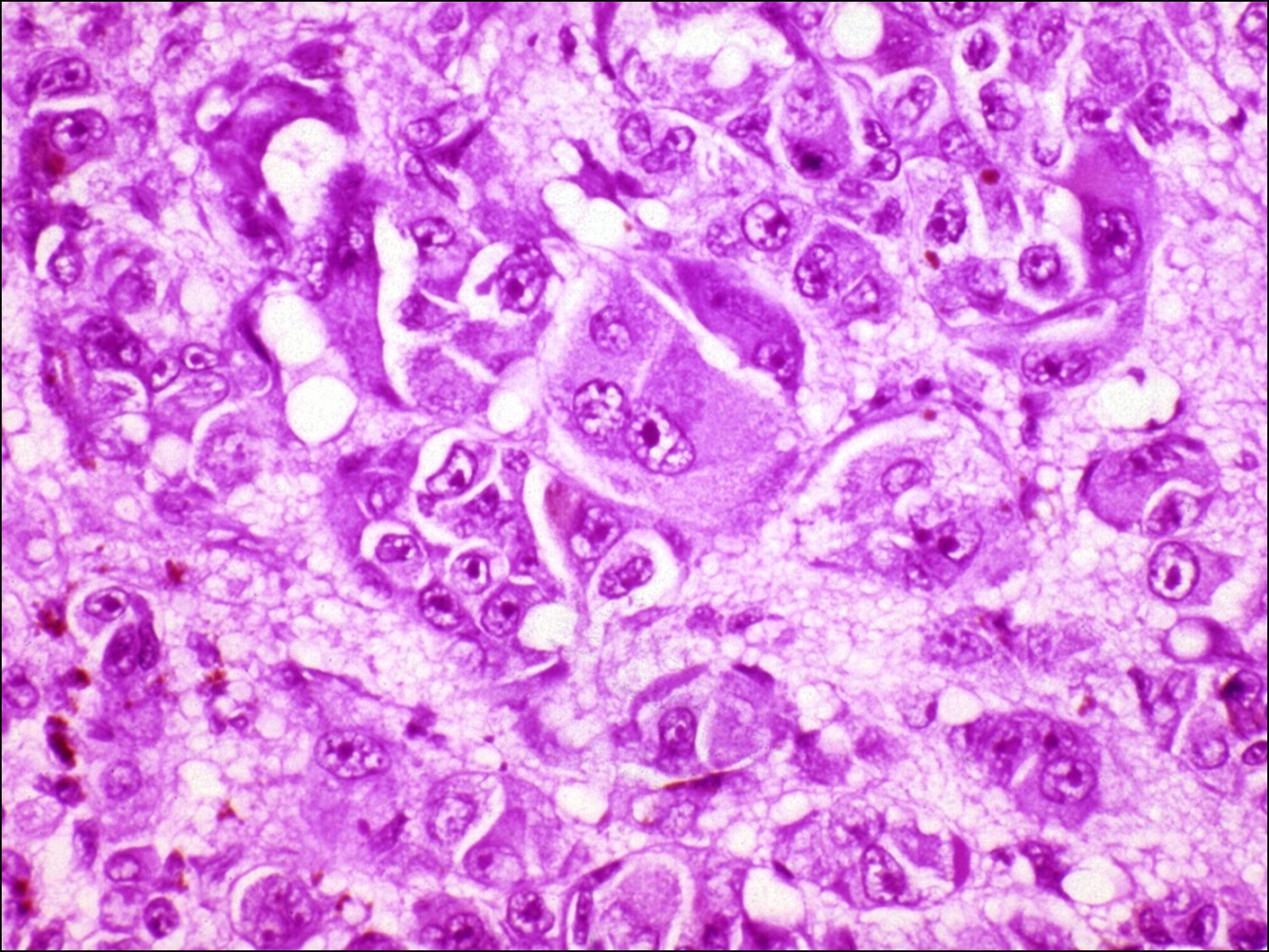
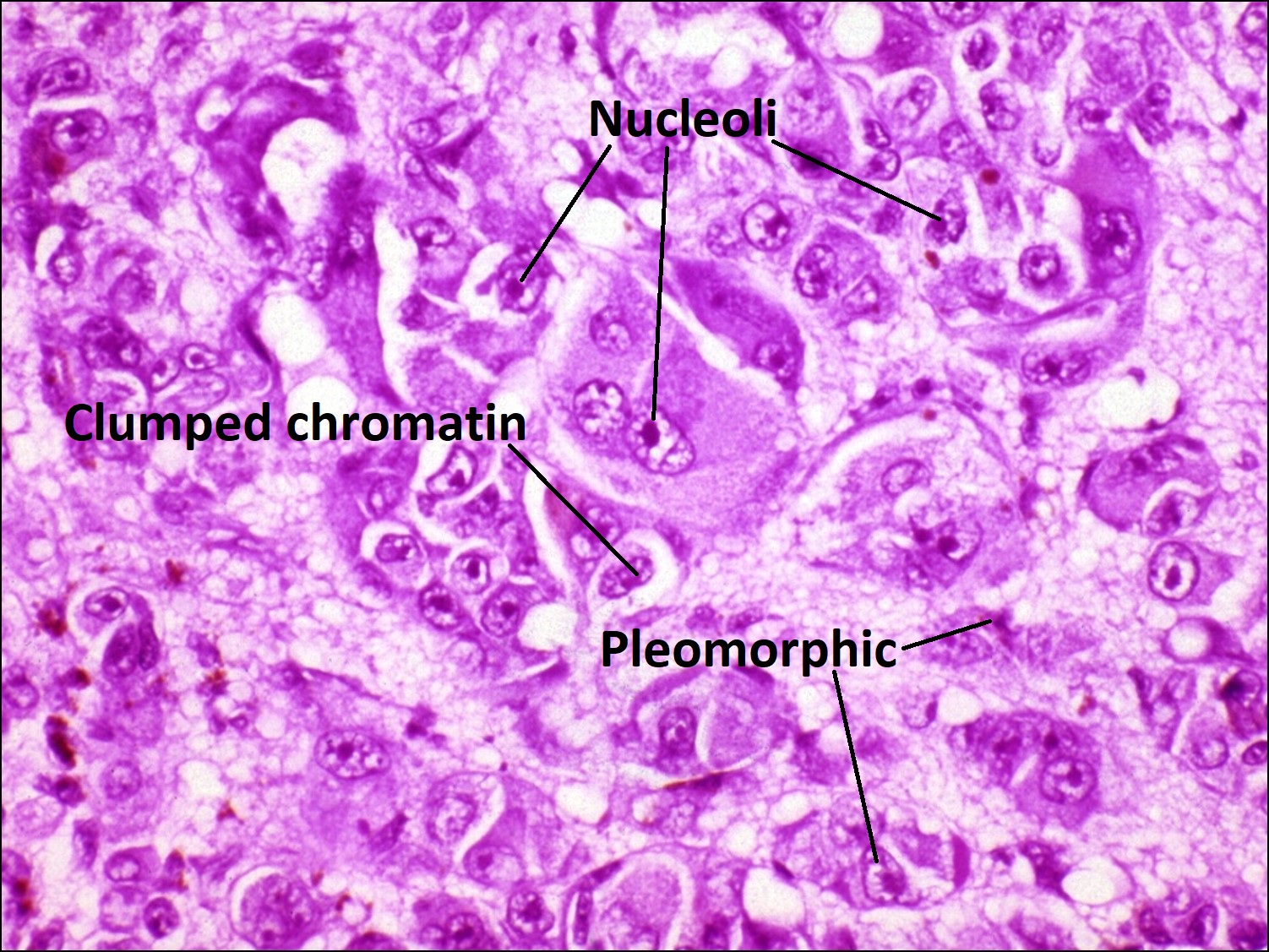
Histology of choroidal melanoma


Low magnification view of choroidal melanoma
Melanoma: most common primary ocular malignancy in adults
- Onset 50-70 years old–much more common in elderly
- 6 cases / million
- Most common in individuals with lightly pigmented eyes
- Nevus of Ota (oculodermal melanocytosis) increase risk
- Ciliary body melanomas may be asymptomatic at 1st–hard to see–some possible signs include:
- May displace lens, may cause sectoral cataract
- May erode through Iris, may make ring melanoma
- Dilated episcleral (sentinel vessels) maybe be present
- Location + size determine presenting features
- Brown dome shaped lesion
- May have mushroom shape if breaks through Bruch’s membrane–mushroom shape makes diagnosis of choroidal melanoma very likely
- Orange (lipofuscin) pigment may be present
- Serous detachment common
- Most important prognostic factors are as follow:
- Size of tumor in contact w/ sclera
- Cell type making tumor
- Spindle cell nevus (not a melanoma)
- Spindle cell melanoma –best prognosis
- Spindle A type –cells have elongated elliptical nucleus with poorly visiblenucleol
- Spindle B type –cells have prominent nucleoli in an oval nucleusc.
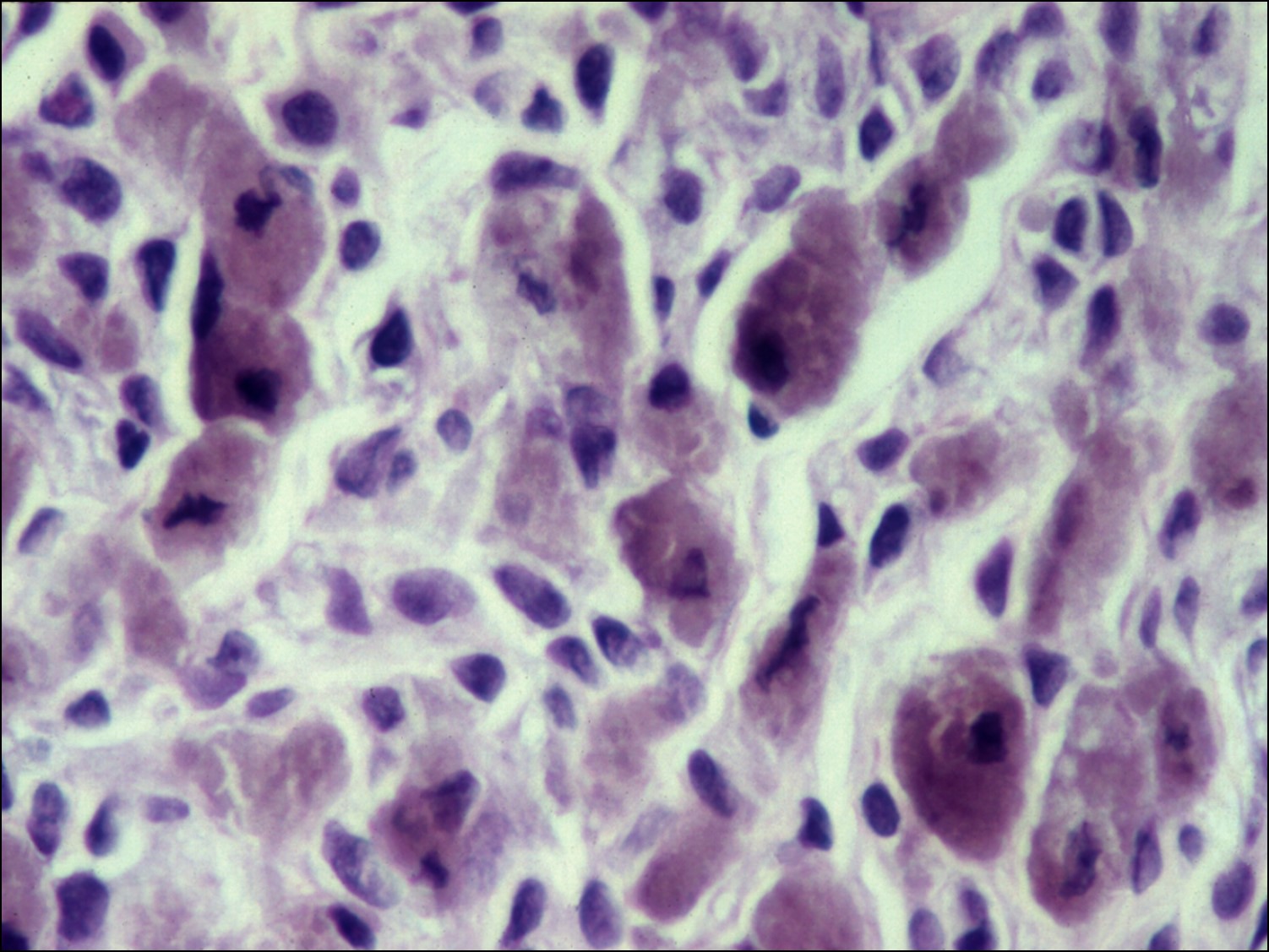
- Epithelioid melanoma
- Worst prognosis
- Least differentiated –superficially resemble epithelial cells
- Polygons with poor adhesion to each other
- Prominent nucleolid
- Mixed melanoma–cells have features of both spindle and epithelioid cells
- Mean of 10 largest melanoma cell nuclei (MLN) has been shown to correlate well w/ survival after nucleation when MLN combined w/ area contracting sclera, then even better
- Complex microvascular patterns have increased risk of metastasis
- Tumor necrosis may incite inflammation
- Pigment liberation may cause secondaryglaucoma (melanomalytic glaucoma)
- Prognosis:
- Spindle A tumors –best survival rate andfatality is rare
- Spindle B tumors–95% survival at 5 years
- Epithelioid containing –5 year survival 58%4.
- Larger tumors have lower survival rates (86% survival for small (<10mm), 66% medium (10-15mm) and 56% for large (>15mm))
- Other poor prognostic factors:
- Increased mitotic figures
- Vascular loops
- Presence of necrosis
- Increased pigmentation
- Anterior location
- Extraocular extension
- Lymphocytic infiltration
- Monosomy 3
- Molecular classification of gene expression in tumor can aid in prognosis
- 95 to 100% of metastasis involve liver
- Types with very poor prognosis include
- Ring melanoma of ciliary body -> very poor prognosis
- Diffuse choroidal melanomas -> poor prognosis
- COMS study: 3 multicenter randomized controlled trials
- Large tumor trial showed no difference in survival between enucleation vs. external beam radiation followed by enucleation
- Medium tumor trial showed no survival difference between I-125 brachytherapy and enucleation
- Small tumor trial showed tumor growth in 21% of tumors by 2 years and in 31% of tumors by 5 years
Metastatic Choroidal Tumors

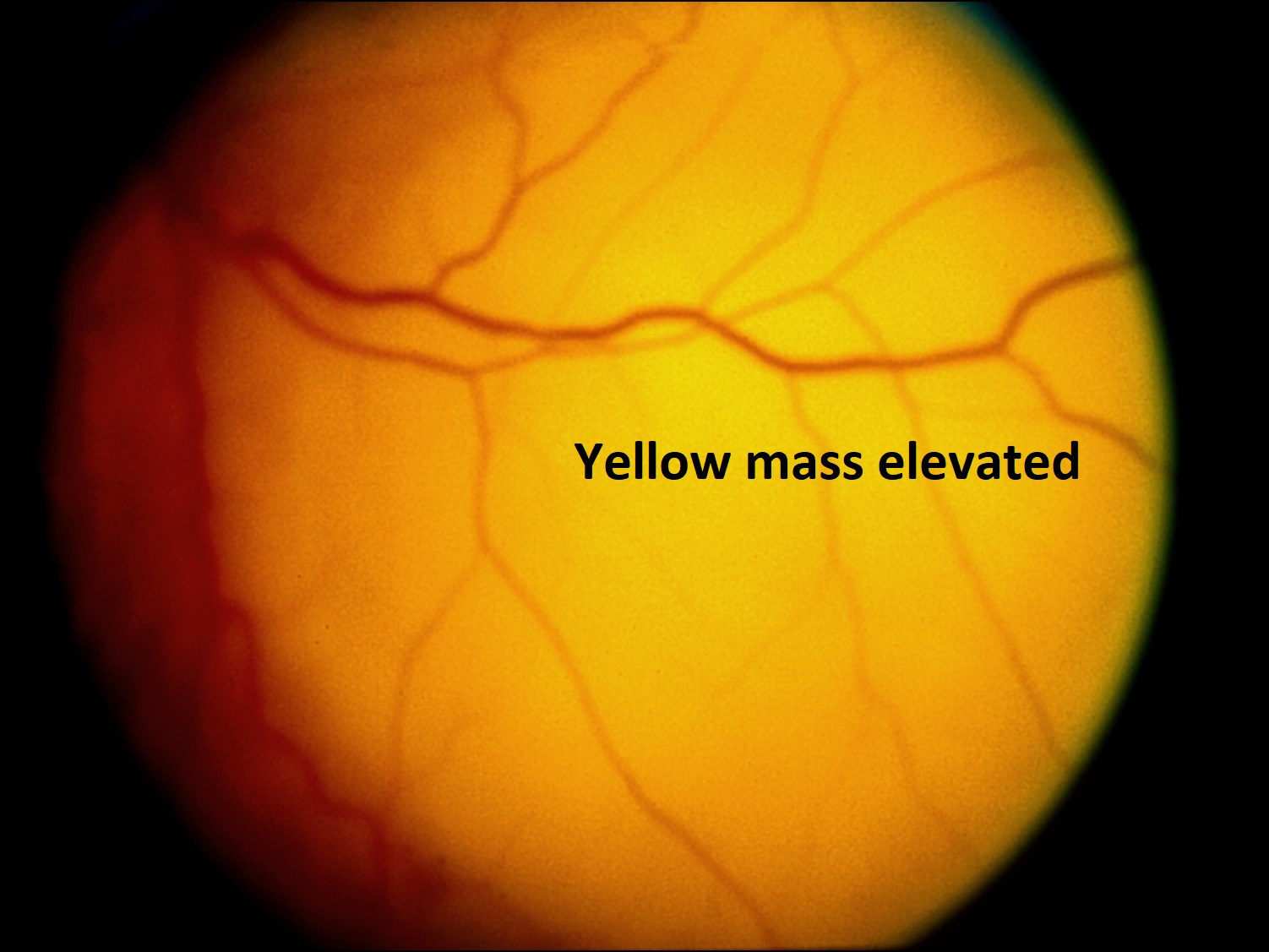
Color photo of metastatic choroidal tumor


Low magnification view of metastatic choroidal tumor
Metastatic Tumors: most common intraocular tumors in adults
- Metastasis most commonly to the posterior choroid (88%)
- May be multiple orbilateral
- #1 in women= breast cancer
- #1 in men = lung cancer
- Poor prognosis –mean survival 9-10 months
Nick’s Tips: usually appears yellow, white or pink mass on fundus exam. Bruch’s membrane almost always intact (mushroom shape is almost always choroidal melanoma). Most breast and lung cancers are mucous secreting so stains for mucin such as alcian blue, mucicarmine, or PAS can identify tumors. Consider immunohistochemistry to identify unknown tumors.
Other Uveal Tumors
Hemangioma:
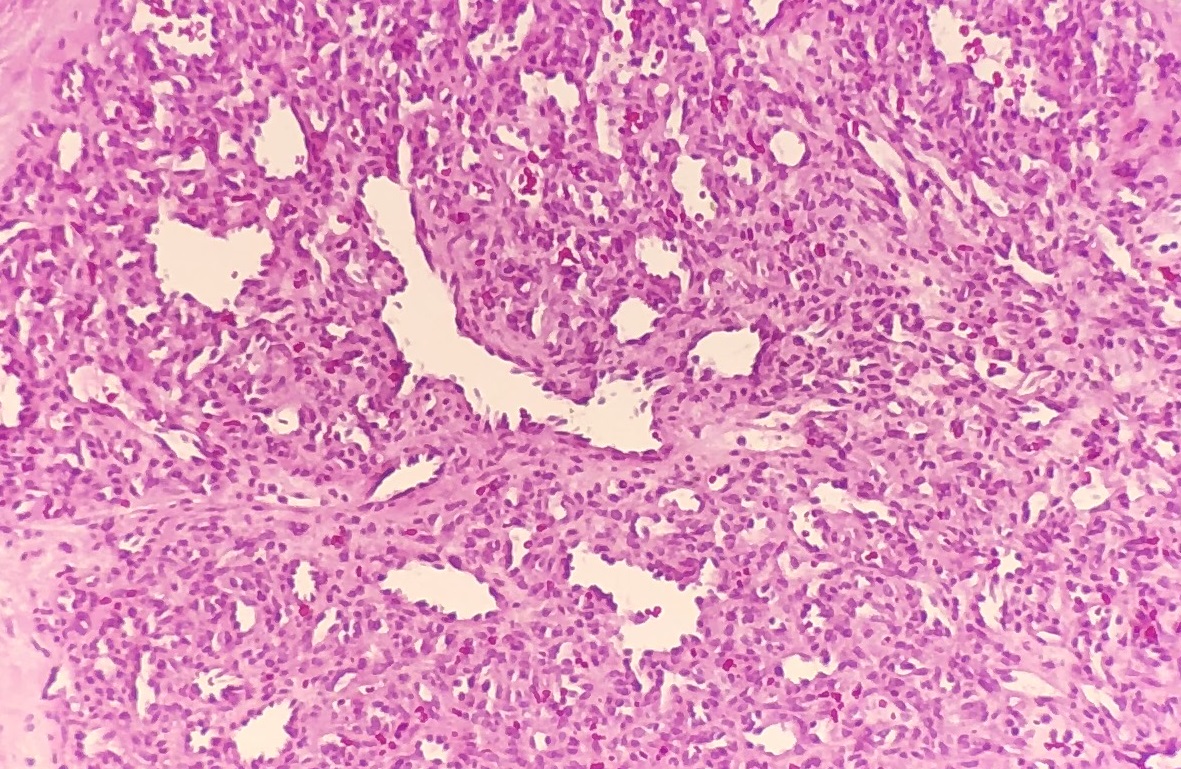

Histology of hemangioma
- Localized
- Not associated with systemic disorders (red orange tumor in post equatorial fundus)
- May cause secondary retinal detachment
- Diffuse –Sturge-Weber Syndrome (encephalotrigeminal angiomatosis)
- Tomato catsup fundus
- Highly reflective on UBM –lots ofchannels and acoustic interfaces.
- Diffuse, thickening of entire fundus –RPE hyperplasia and metaplasia may occur
- Blood vessels (unlike orbital cavernous hemangioma) show very little intervening stroma
- Associated with serous retinal detachment
- May lose vision due to chronic CME or detachment or glaucoma (present in about 75% of eyes)
- Glaucoma may be due to
- Anterior displacement of iris diaphragm with pupillary block and angle closure
- Malformation of the angle
- Iris and angle neovascularization
- Elevation of episcleral venous pressure
- Treatment:
- Observe
- Laser around tumor to decrease likelihood of serous retinal detachment
Choroidal Osteoma:
- Benign bony tumors (compact bone)
- Adolescent to young women > men
- Yellow/orange with well-defined margins
- May slowly enlarge, impairing vision if it involves the macula
- Often bilateral and peripapillary
- Very high reflectivity on ultrasound and very bright on CT
Nick’s Tips –Look for compact bone located within the choroid on histopathology –in phthisis, the bone is located on the inner surface of the choroid associated with the RPE.
Neurofibromas


Histology of neurofibroma
- Benign peripheral nerve sheath tumor that usually arise sporadically but can be inherited in association with neurofibromatosis type 1
- Appear in 2ndto 3rddecades of life and affect men and women equally
- Usually present as slowly growing, skin-colored, rubbery nodules that are evenly distributed superficially throughout the body surface
- Histology shows a prominent proliferation of wavy, elongated
- Schwann cells w darkly staining nuclei interspersed with bundles of “shredded carrot” collagen. All other peripheral nerve components may be present as well.
- Strong CD34 and S100 positivity on immunohistochemistry
Schwannomas


Schwannoma, Antoni A form, which is a noted by the spindle-shaped cells which line up in a palisading pattern.


Schwannoma, Antoni B form, which has less organized, myxoid appearance
- Also known as neurilemomas
- Benign peripheral nerve sheath tumor from differentiated Schwann cells
- Most schwannomas arise sporadically but can be inherited in association with neurofibromatosis types 1 and 2
- Appear in 3rd to 6th decades of life and affect men and women equally
- Usually located on limbs, head, neck or orbit
- Histology shows a biphasic pattern of Antoni A and Antoni B areas
- Antoni A-hypercellular areas of compact, spindled cells
- Antoni B-hypocellular, myxoid areas with macrophages and collagen
- Strong, diffuse S100 positivity on immunohistochemistry; also positive for vimentin and CD68
Leiomyoma:
- Rare tumors from smooth muscle of ciliary body displaying myogenic and neurogenic features
- Usually in young women
- Smooth muscle actin and muscle markers differentiate leiomyomas from schwannomas and melanomas
Nick’s tips: paucicellular tumor from the ciliary body area –consider schwannoma or melanoma in differential, but smooth muscle markers can make the diagnosis
Lymphoid Proliferation/Primary Choroidal Lymphoma:
- Masses of well-differentiated, small lymphocytes
- Similar to spectrum of low-grade lymphoid lesions
- Usually MALTOMA, germinal centers are rare
- Polymorphic lymphocyte proliferation
- Lymphoma in the uveal tract is possible
Trauma of the Choroid:
- Prolapse of uveal tissue through scleral rupture after penetration is common
- CNV may occur as late complication
- Choroidal detachment: hemorrhagic or serous fluid between choroid and sclera. May require surgical drainage
- Chorioretinitis Sclopetaria: traumatic rupture of choroid and retina
Nick’s Tips: Remember trauma may be non-accidental (child abuse). The pathologist may play a role in proving trauma was abuse and careful examination of tissue may be an important factor in court cases to establish cause of death or injury.