
Intraocular Tumors – Melanocytic
Home / Ophthalmic Pathology / Intraocular Tumors
Differential Diagnosis of iris nodules
- Cyst, Epithelial invasion
- serous or solid cysts following surgery
- Trauma
- Retained foreign body -usually secondary pigmentation of iris → chronic iridocyclitis and PAS
- Fungal endophthalmitis -Irregular yellow-white mass, cell in AC, +/-hypopyon
- Iridocyclitis -Granulomatous nodules –superficial or deep–associated with
- sarcoidosis
- Koeppe nodules located at the pupillary border
- Busacca nodules located on anterior iris surface
- Iris freckle -stationary, light-dark, flat, anterior, with increased pigment, but no hyperplasia
- Iris Nevus -discretemass on anterior iris surface
- Composed of benign nevus cells.
- Increased incidence in neurofibromatosis-1
- Cogan-Reese iris nevus syndrome
- Acquired diffuse nevus: associated with
- Glaucoma
- Heterochromia
- PAS
- ICE syndrome
- Iris pigment epithelial cysts -encompass both layers of iris and produce localized stromal elevation. May need B-scan or trans-illumination to see.
- Iris pigment epithelial proliferation results from congenital or acquired (trauma/surgery) –composed of plaques of pigment epithelium –black, velvety color.
- Juvenile xanthogranuloma -yellow/grey iris lesions with orange skin
- Lesions in 1st yearof life.
- Assoc. 1. with spontaneous hyphema
- Secondary glaucoma
- Diffuse granulomatous reaction with lipid filled histiocytes and touton giant cells Regress spontaneously
- Leiomyoma
- localized or diffuse, pedunculated or flat
- Electron microscopy required to differentiate from amelanotic spindle cell melanoma
- Leukemia -very rare
- Nodularor diffuse milky lesions with intense hyperemia.
- Pseudohypopyon
- Iris loses architecture and becomes thickened.
- Lisch Nodules
- one of the diagnostic criteria for NF-1
- Multiple flat or raised tan to brown lesions
- Composed of collections of nevus cells
- Malignant melanoma
- Nodular of flat –usually peripheral
- 80% inferior + inferotemporal
- May have nutrient vessel, satellite pigmentation
- Pupil may dilate irregularly
- Melanocytosis
- Unilateral, heterochromia irides
- May have oculodermal form with eyelid + brow involved
- Has malignant potential
- Retinoblastoma
- White foci on iris surface or in angle
- May have pseudohypopyon
- Retinoblastoma present in posterior chamber
- Tapioca Melanoma
- Often associated with unilateral glaucoma.
- Tapioca-like nodules over part or all of iris
- Metastatic Carcinoma
- Gelatinous to white vascularized nodules on iris surface.
- May be associated with anterior uveitis, glaucoma, rubeosis, and hyphema.
Iris Nevus:
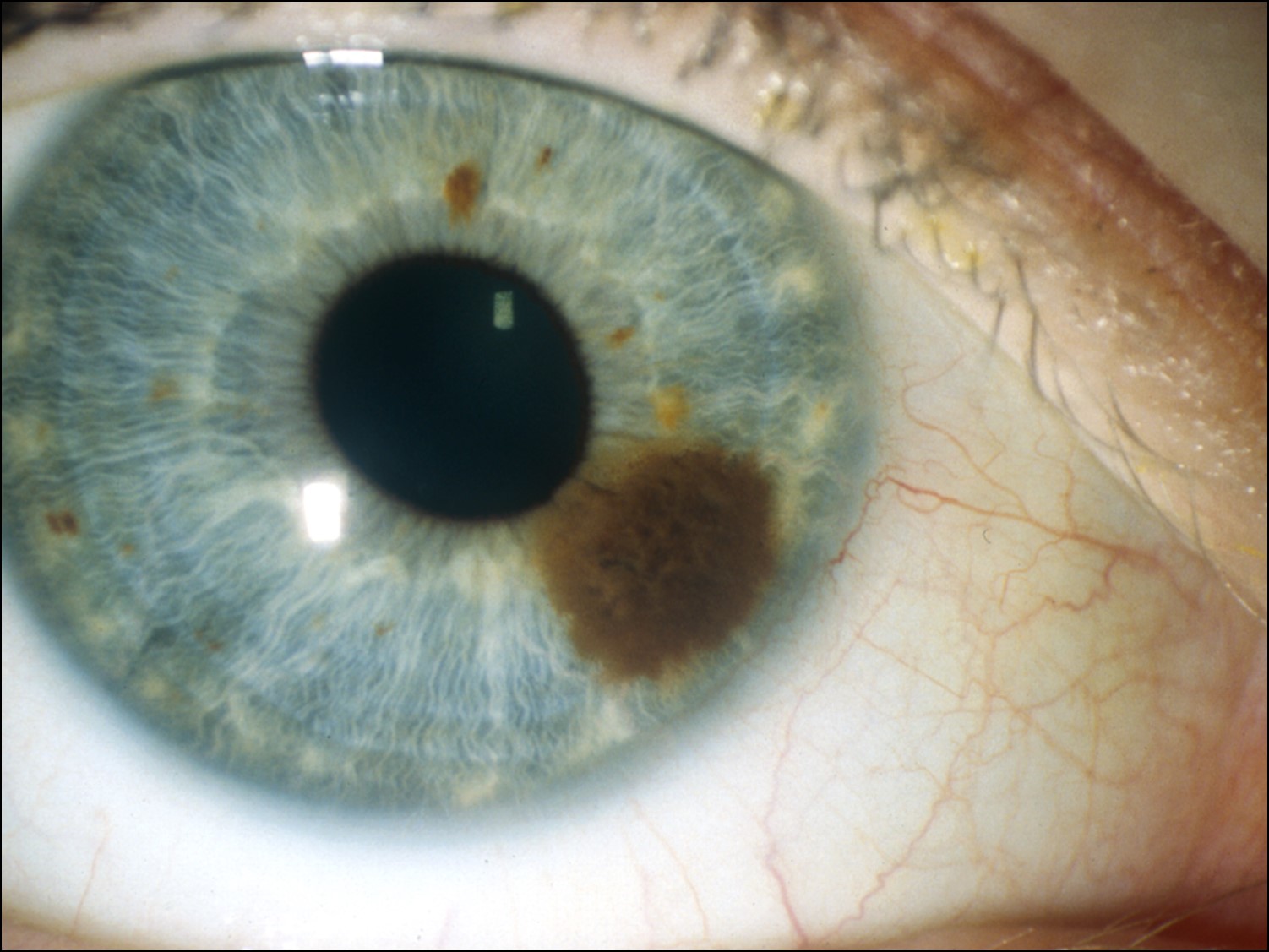

17.01 External slit lamp photo

17.02 Spindle nevus


17.03 Borderline spindle nevus

17.04 Epithelioid nevus
- Darkly pigmented lesion of iris stroma
- minimal distortion of normal architecture
- circumscribed > typically nodular + discrete
- diffuse > may involve entire sector or even entire iris
- increased incidence in NF –1
- most common in Caucasian background (97%)(1)
- ABCDEF guide for risk factors 1–risk of nevus transformation to melanoma increases for the following:
- A –age –younger age is higher risk
- B –blood –hemorrhage is common in melanoma
- C –clock hour inferiorly –inferior location is higher risk
- D –diffuse configuration –diffuse nevus more likely for malignant transformation
- E–ectropion –ectropion uvea increases risk
- F –feathery margin
Iris Melanocytoma:
- Dark pigmented brown lesion –may have granular “mound of black sand” appearance.(1)
- Usually stable and does not require intervention
- May have spontaneous necrosis –which can liberate pigment into anterior chamber and cause glaucoma.
- In one series of 47 patients, 48% showed mild growth and 63% showed new tumor seeds, but none (0%) showed malignant transformation(4).
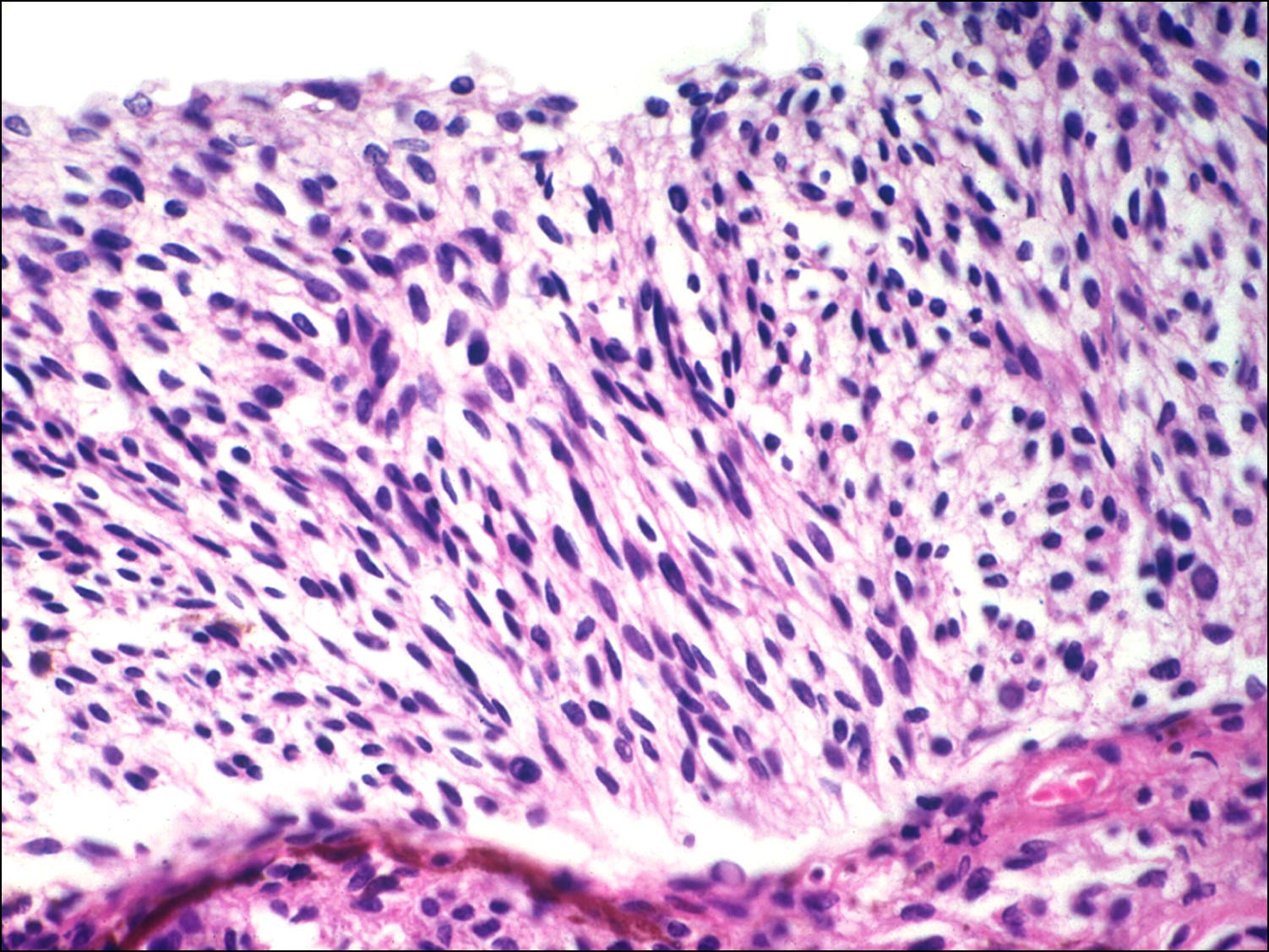
Iris Melanoma:


17.05 External slit lamp photo


17.06 Spindle melanoma

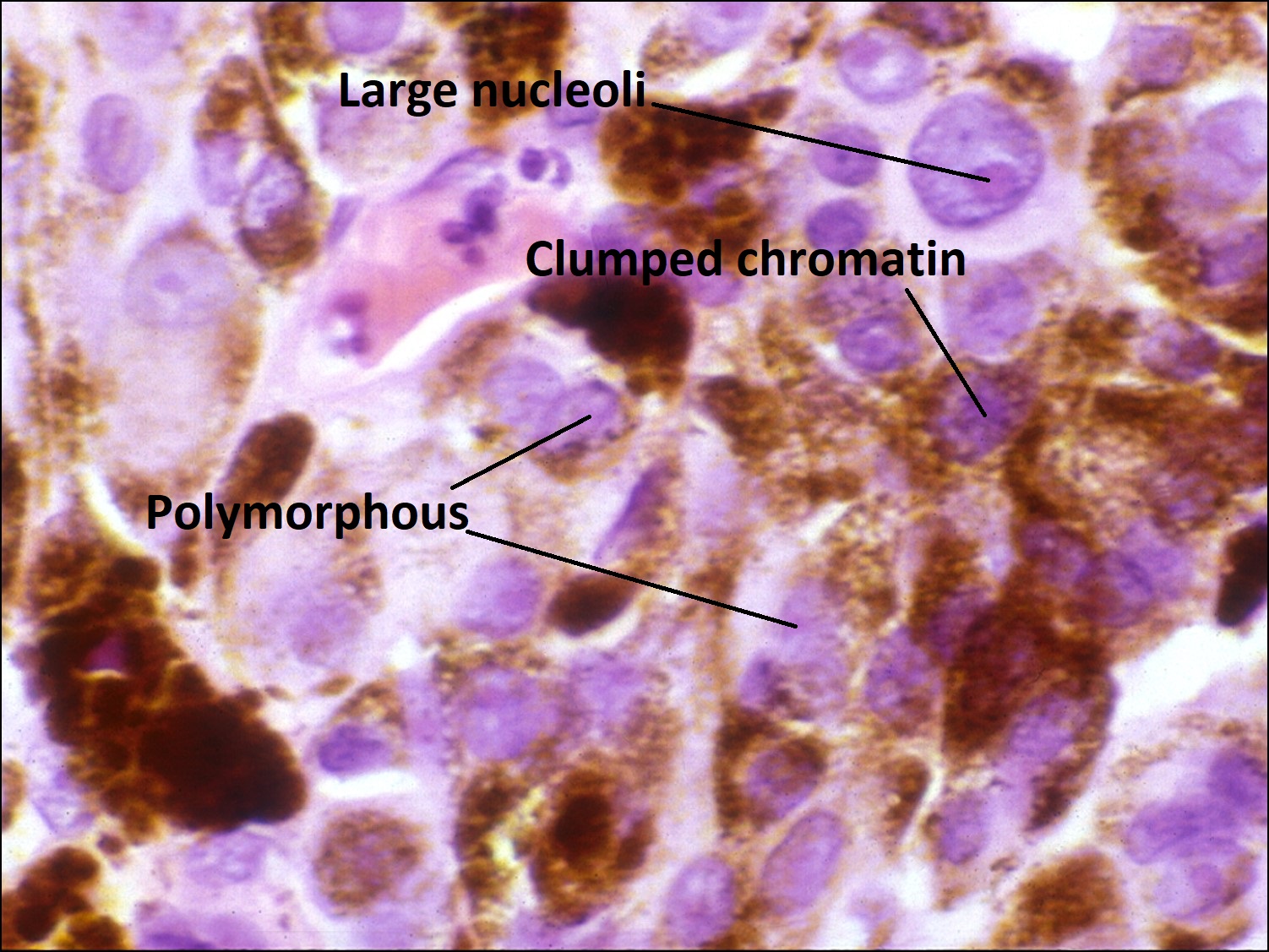
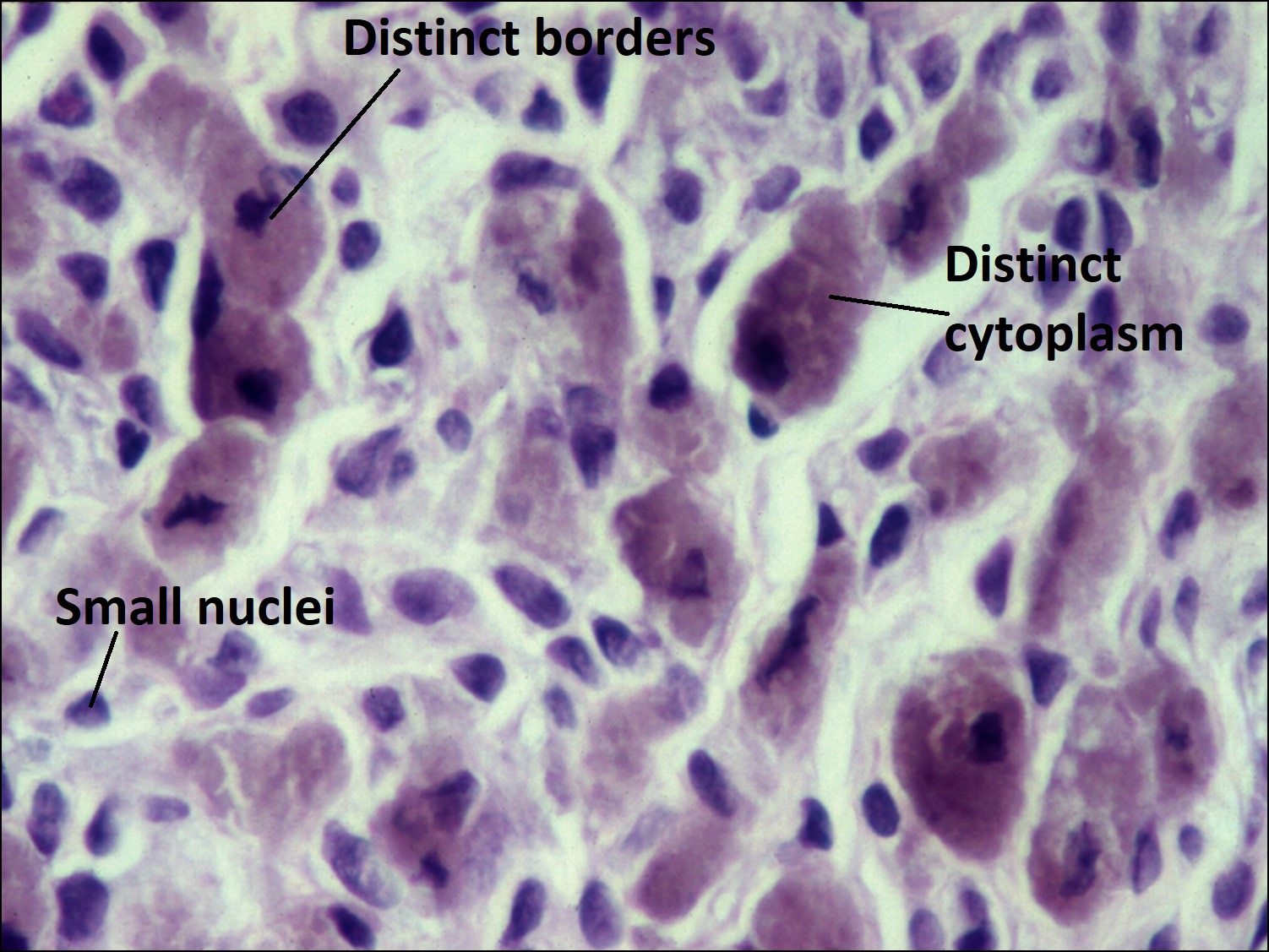
17.07 Epithelioid melanoma
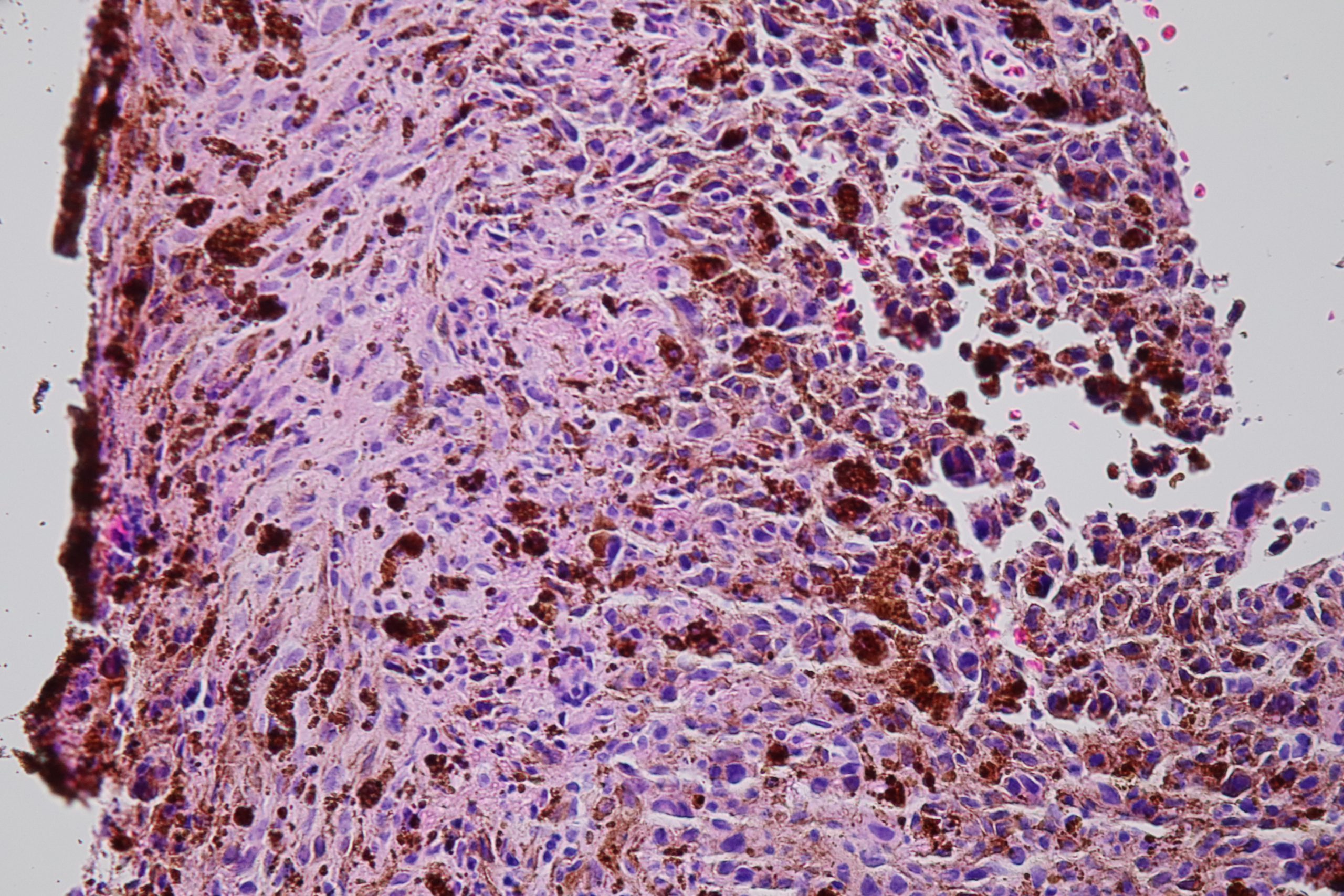

17.08 Mixed melanoma
Nick’s Tips: When small iris melanoma may be impossible to differentiate from nevus –may consider biopsy done through clear cornea.
- Risk of metastasis is about 3%,(5) much less commonly fatal than other choroidal melanomas possibly due to
- Earlier diagnosis and usually smaller at diagnosis
- Most commonly spindle cell type(over 50%)
- May have other unique features
- Least common site of primary uveal melanoma (about 3-10%).(6)
- Associated with: (6)
- Light iris color
- Inferior location
- Average age 40 years old (younger than choroidal melanoma (age 50))
- May appear circumscribed (melanotic nodule) or diffuse (heterochromia), with diffuse having a somewhat poorer prognosis.
- Histopathologic features:
- Typically spindle uveal melanoma cells –
- Plump nucleus,
- Eosinophilic nucleolus visible,
- Mildly coarse chromatin
- Typically spindle uveal melanoma cells –
- Some are mixed cell type (combination of spindle and epithelioid
- Epithelioid –
- Abundant eosinophilic cytoplasm and distinct cell borders
- Large nucleus
- Central nucleolus
- Larger and more pleomorphic than spindle cells.
- Signs of iris melanoma:
- Ectropion iris
- Prominent Vascularity
- Sectoral cataract
- Secondary glaucoma
- Progressive growth
- Extrascleral extension
- Seeding of angle
- Large size
- ¾ of iris melanomas involve inferior iris
- Unilateral glaucoma
- Spontaneous hyphema
- Treatment
- Excision
- Brachytherapy
- Proton beam
Iris Cysts:

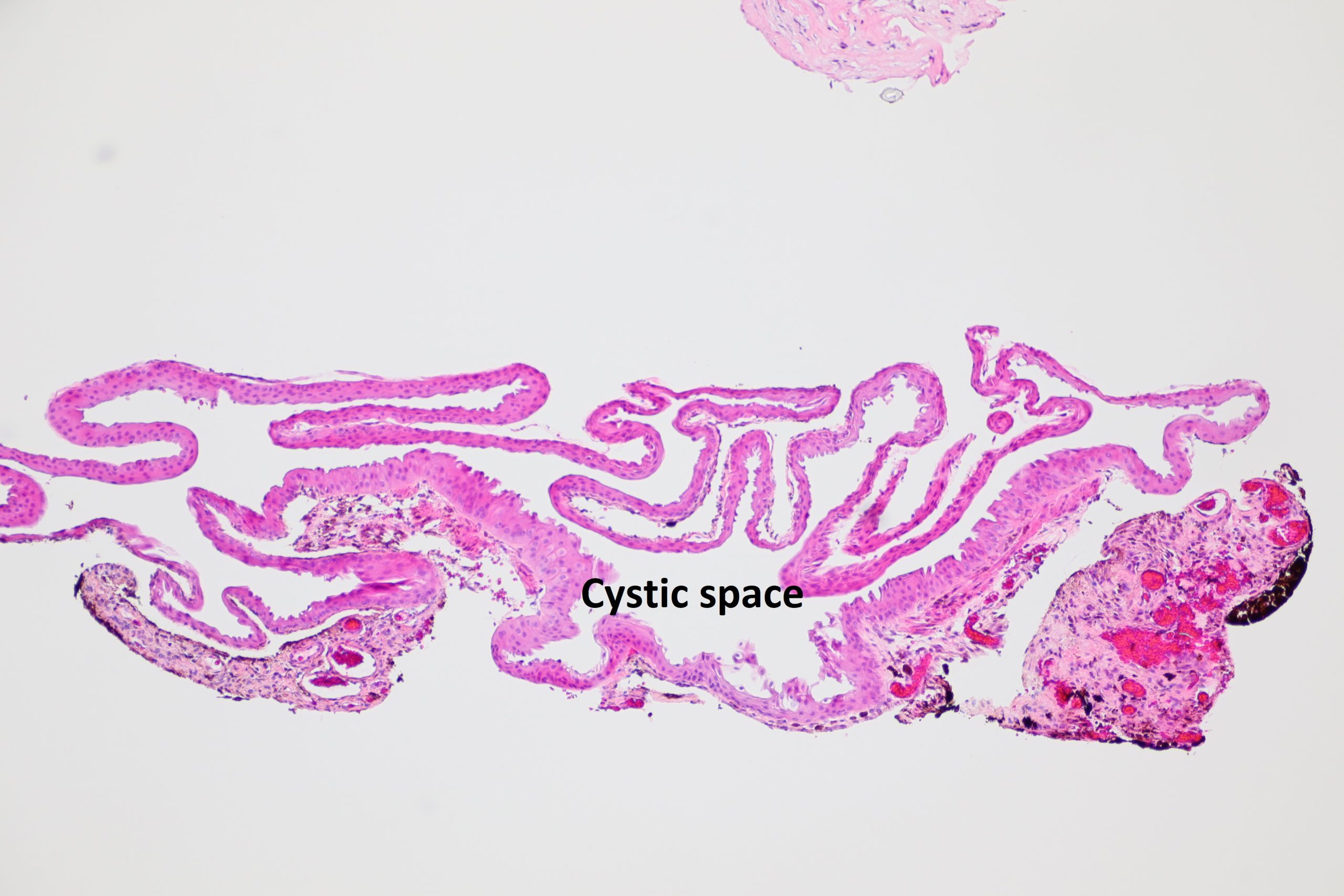
17.09 Iris stromal cyst
- Imaging:(2)
- Ultrasound (UBM) –better at imaging posterior wall and overall imaging of structure
- Anterior Segment OCT –better at imaging anterior surface of lesion
- Types of iris cysts
- Stromal cysts
- may be congenital or acquired
- usually smooth, translucent mass on in in the iris
- may have debris inside cyst
- may rupture –can cause iritis or glaucoma if ruptured
- Congenital stromal cysts in children under age 10 tend to be more aggressive and may have poor visual outcome.
- Treatment –may surgically remove or may aspirate cyst and sclerose with absolute alcohol.(3)
- Pigmented epithelial cysts
- Usually asymptomatic
- Darkly pigmented –arising from posterior surface of the iris
- May resemble ciliary body melanoma or iris pigmented epithelial adenoma, clinically.
- UBM and anterior segment OCT may help demonstrate cystic nature of lesion.
- Stromal cysts
Nevus of choroid / ciliary body


17.10 Fundoscopic photoof nevus
Nick’s Tips: Although no single feature differentiates a malignant melanoma and a choroidal nevus, virtually all melanocytic tumors greater than 3mm thick are melanomas and virtually all melanocytic lesions less than 1mm thick are Nevi.
- Choroid nevus –7% of population –flat or minimally elevated
- May be amelanotic or grey-black
- Differential diagnosis of a pigmented fundus Lesion
- Nevus
- Melanoma
- Atypical disciform scar
- Suprachoroidal hemorrhage
- RPE hyperplasia
- CHRPE
- Melanocytoma
- Choroidal osteoma
- Hemangioma with RPE changes
- Metastatic tumor with RPE changes
- Risk of melanoma
- Increased with thickness > 1 mm
- Increased with diameter > 10mm
- (<10mm almost always benign)
- Visual symptoms
- Orange pigment
- Subretinal fluid
- Large size >1mm thick, >10mm diameter
- Juxtapapillary
- Absence of drusen or RPE changes
- Hot spots on FA
- Homogeneity on ultrasound
- Enlargement
Melanocytoma:


17.11 Ciliary body melanocytoma
- Rare, large polyhedral shaped cells with small nuclei + melanin.
- May seed A/C causing glaucoma
- In choroid, they appear like nevis or melanoma on exam
- May undergo malignant changes.
- If growth is documented, treat as malignant.
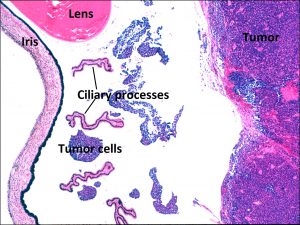
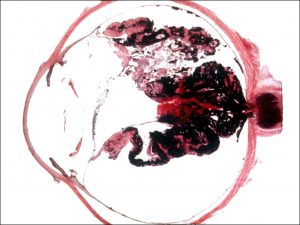
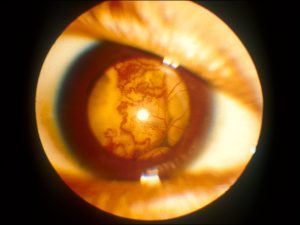
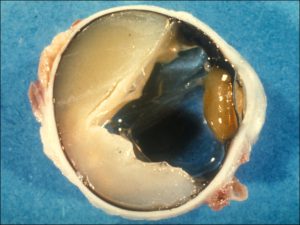
Melanoma of ciliary body/choroid:


17.12 Ciliary body melanoma

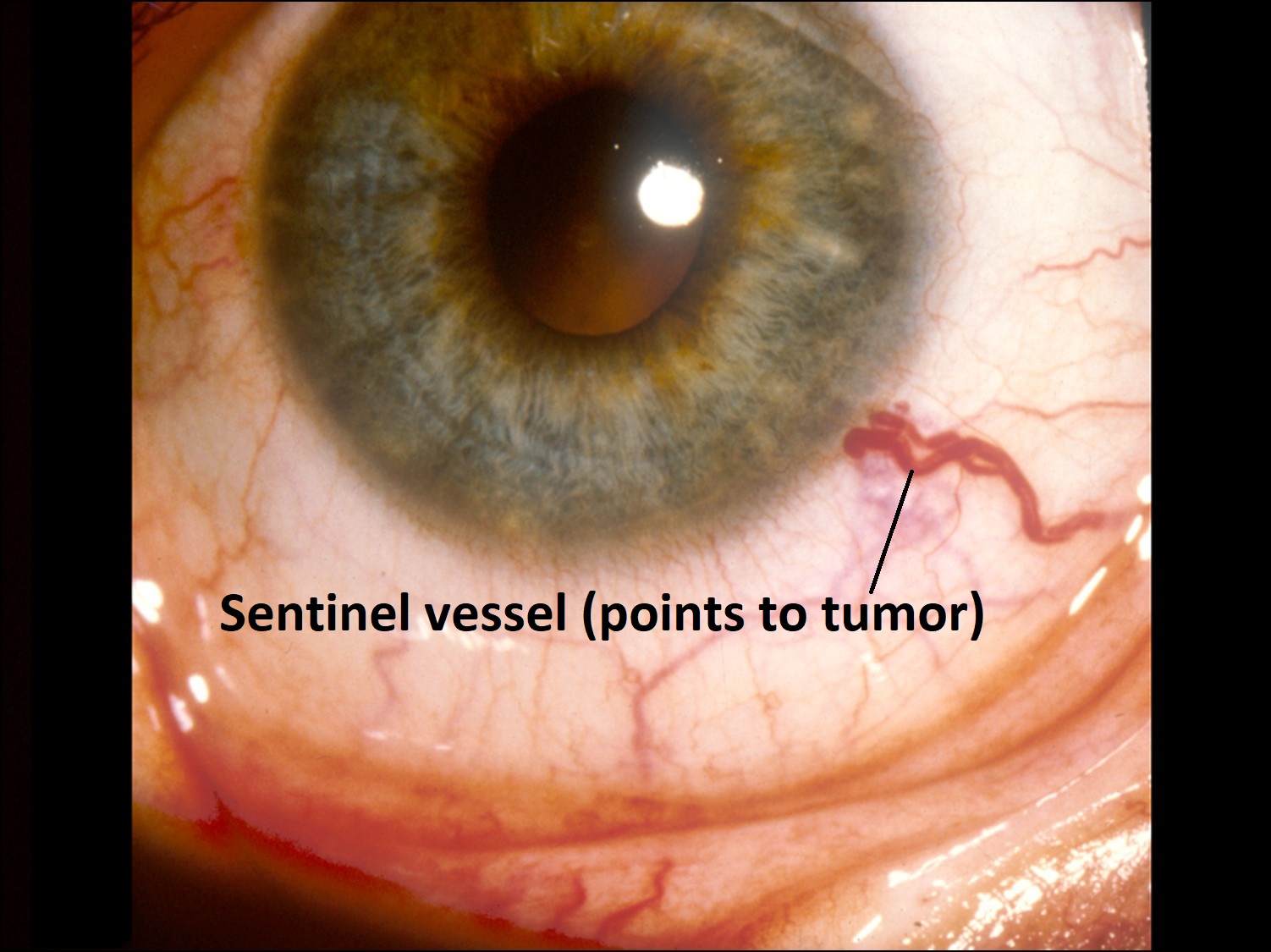
17.13 External slit lamp photo

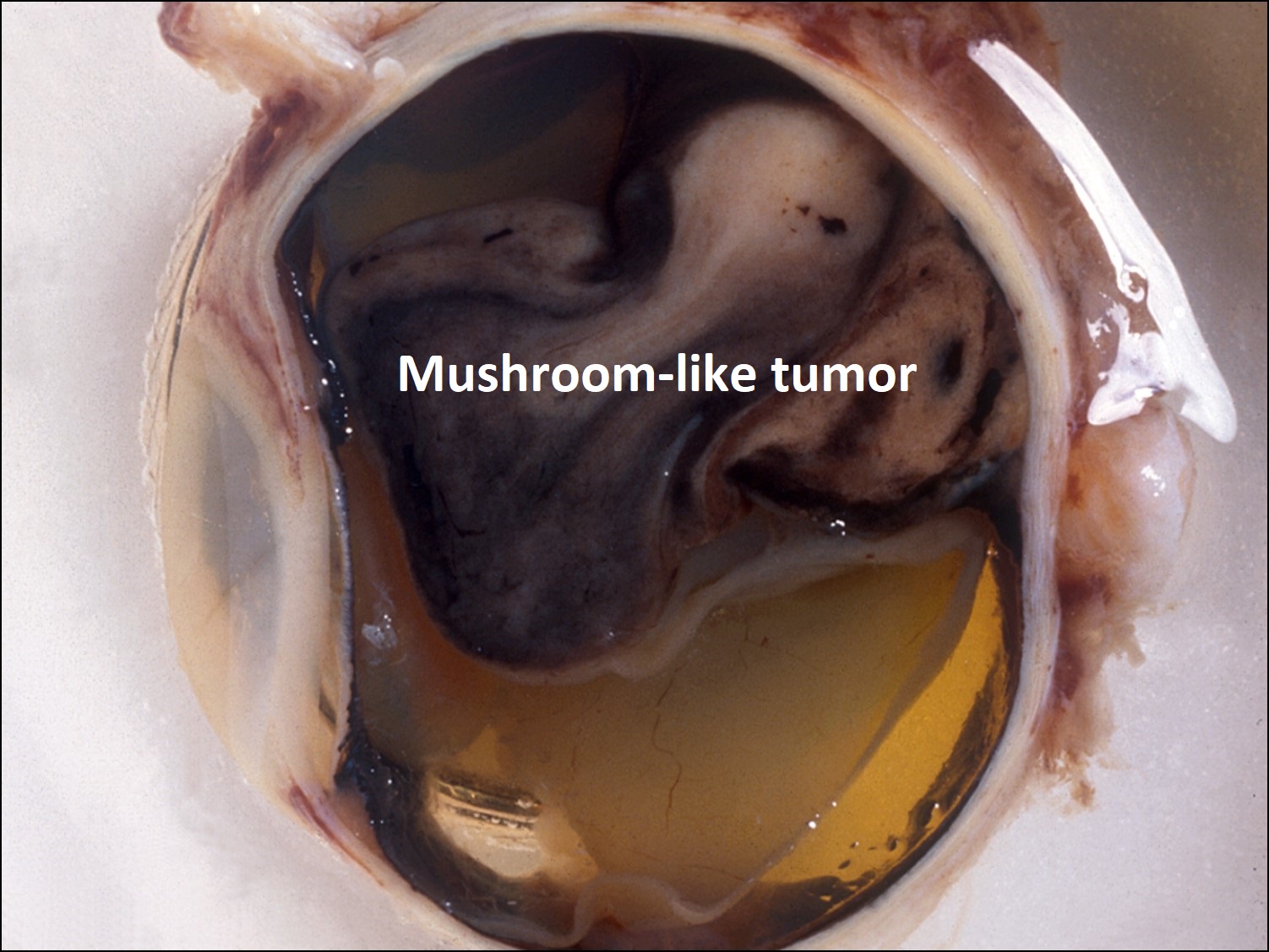
17.14 Gross cross-section of globe


17.15 Tumor arising from ciliary body

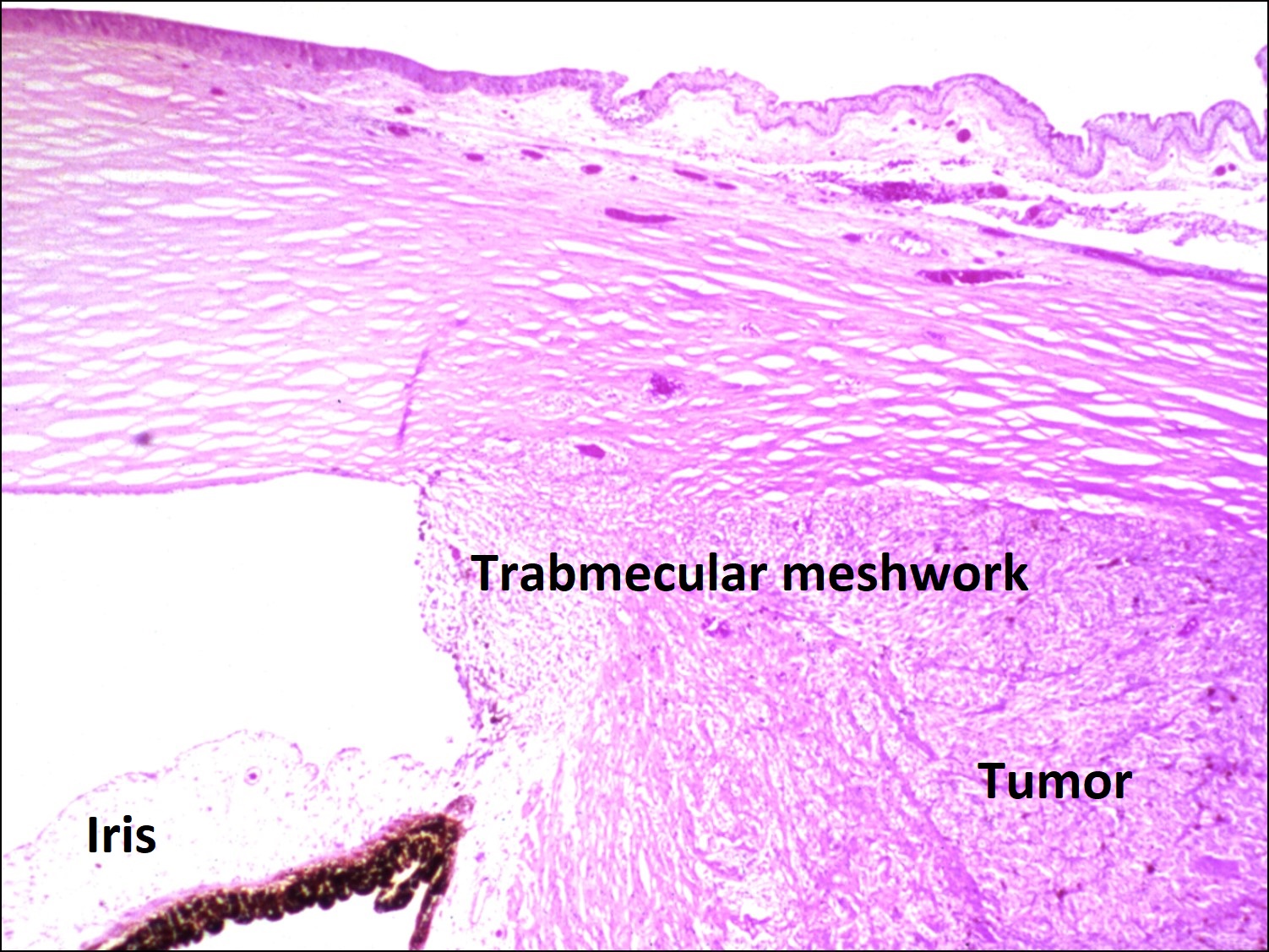
17.16 Tumor arising in ciliary body coming into trabecular meshwork


17.17 Choroidal Melanoma

17.18 Spindle cell A choroid
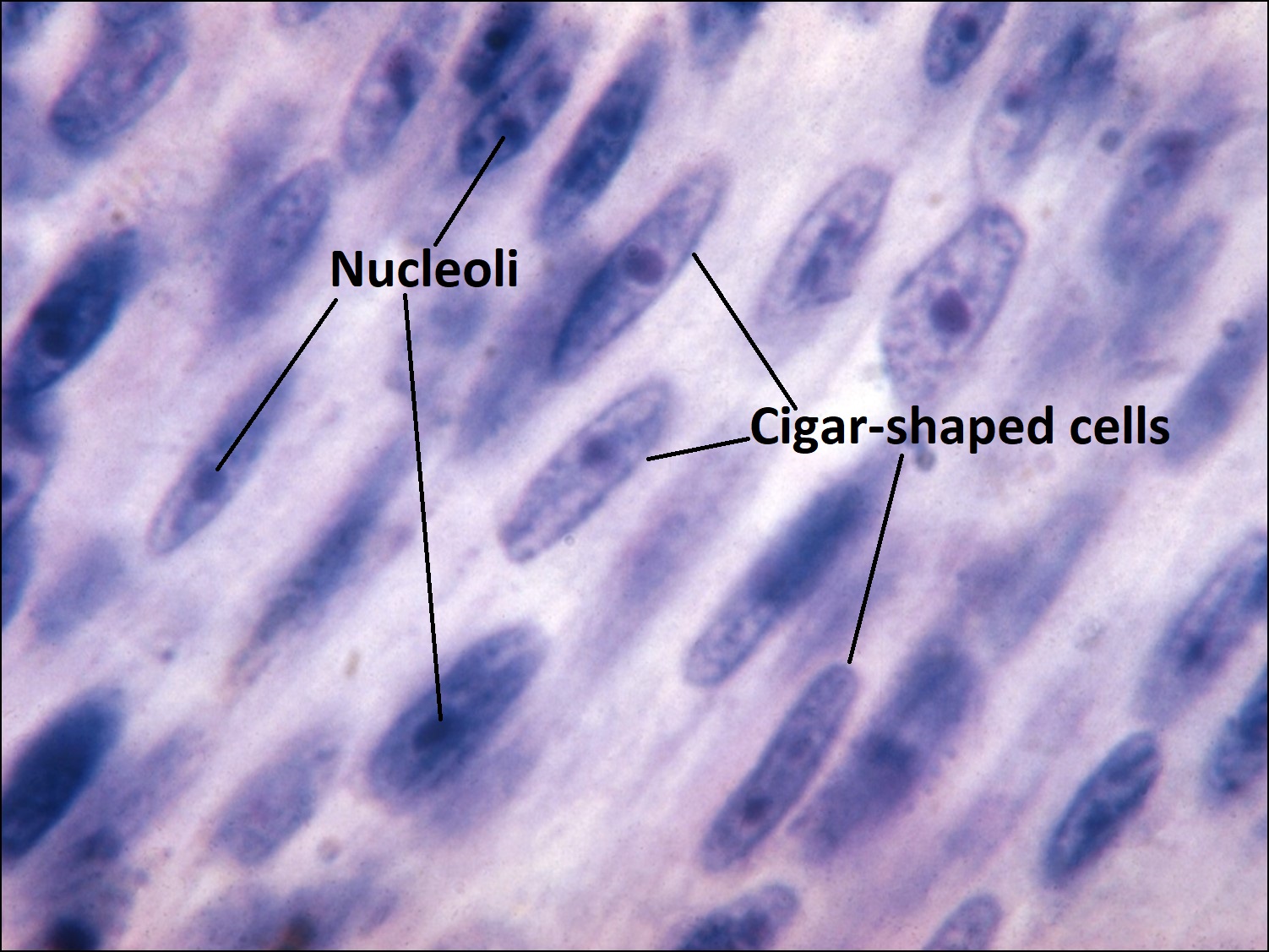
17.19 Spindle cell B choroid
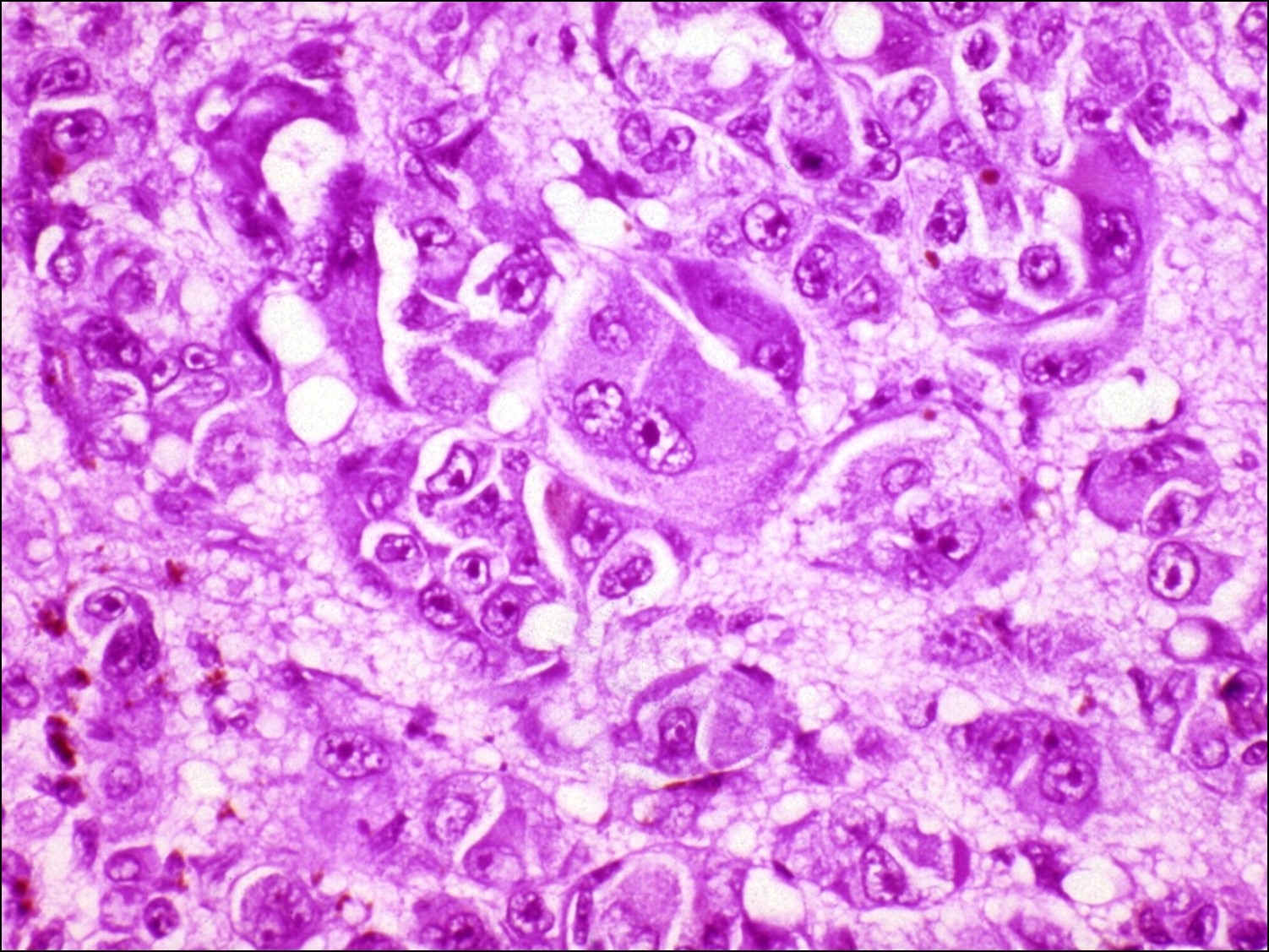

17.20 Epithelioid choroid


17.21 Mixed melanoma choroid
Nick’s Tips:
Histopathology of Choroidal Melanoma/Nevus:
- Six types:
- Spindle nevus
- Spindle nevus with plaque
- Borderline spindle nevus
- Spindle melanoma
- Epithelioid nevus
- Epithelioid melanoma
- Prognosis from histological features –poor outcomes due to metastasis via scleral emissary channels
- Pure spindle B –good prognosis
- Mixed is intermediate prognosis
- Epithelioid is poor prognosis
Clinical Features of Choroidal Melanoma
- Most common primary intraocular malignancy in adults 6-7 cases per million.
- Primary affects patients aged 50’s-60’s
- Risk factors:
- Light complexion, light irides
- Ocular melanocytic conditions
- Melanosis oculi, oculodermal melanocytosis
- Genetic predisposition (dysplastic nevus syndrome)
- Cigarette smoking
- May be clinically silent + large when detected.
- Symptoms may include
- Vision loss or decreased vision
- Changes in or decreased visual field
- Photopsias
- May have no symptoms
- Appearance:
- May rarely erode through sclera or iris + become visible on external exam
- Initial sign of ciliary body melanoma may be dilated episcleral sentinel vessels.
- Ring Melanoma → 360° -180° melanoma of ciliary body.
- Pigmented, elevated dome shaped.
- May be amelanotic to dark brown.
- May erupt through Bruch’s membrane + make mushroom shape.
- Orange pigment clumps may form @ RPE level.
- Serous detachment of retina is common
- Neovascularization, hemorrhage, lens subluxation are late complications
- Vitreous hemorrhage only if penetrated through Bruch’s
- Diagnostic Evaluation:
- Indirect ophthalmoscopic evaluation-#1 technique.
- Gonioscopy –best method for establishing anterior involvement
- High-frequency ultrasound for iris + ciliary body evaluation
- B-scan u/s →for tumor measurement, extrascleral extension
- A-scan u/s →for internal reflectivity (characteristic low internal reflectance –homogenous)
- Transilluminationmay help identify tumor
- Wide angle fundus photography.
- FA does NOT have characteristic appearance.
- CT + MRI may help in opaque media
- Differential diagnosis:
- choroidal nevus: usually flatter + smaller
- ARMD with pigmented scar –fluorescein angiographypathognomic –hemorrhage rare in melanoma
- CHRPE –well defined, flat dark lesion-histology → tall melanin containing RPE cells that are histologically identical to bear tracks.
- Melanocytoma of the optic disc.
- Has minimal malignant potential
- Lesion is located peripapillarily.
- 10% will show growth over 5 years
- May produce vision and visual field defects due to nerve fiber layer disruption
- Suprachoroidal Detachments:
- May be hemorrhagic orserous.
- Associated with hypotony.
- Often dome shaped, associated with breakthrough vitreous hemorrhage.
- Usually located in multiple quadrants
- Usually treated with observation
- Choroidal Osteoma:
- Benign bony tumors. Yellow to orange, well defined
- Arise from juxtapapillary choroid
- Adolescent to young adults
- High amplitude echo on u/s from bony plate with shadowing behind lesion.
- Calcification on CT scan.
- Typically slow growing over years
- May affect vision if in macula or nearby
- Etiology unknown, may be related to low-grade inflammation
- Subretinal neovascularization common
Classification of Melanomas:
| Nervus | <5mm diameter | <2mm thick |
| Small | 5-10mm | 2-3mm thick |
| Medium | 10-15mm | 3-5mm |
| Large | 15-20mm | 5-10mm |
| Extra Large | >20mm | >10mm |
- Metastasis up to 50% at 25yrs, 25% at 5yrs, 34% at 10yrs (COMS Study)Less than 2% have clinically detectable metastatic disease at time of diagnosis. Median survival less than 6months with metastatic disease.Liver 1st site of metastasis, 89% on clinical exam 100% on autopsy. Lung, 24% on clinical exam 50% on autopsy; skin, 12% on clinical exam 50% on autopsy; bone possible later 17% on clinical exam 50% on autopsy.
- Clinical Evaluation: (yearly follow-up)
- Liver ultrasound
- Liver function tests (LFTs)
- Chest X-ray (low yield)
- If any are positive tests, then Tri-phasic liver CT, CT or MRF of chest + abdomen.
- Metastases can develop up to 20 years after treatment.
- Treatment: little evidence of natural history exists.
- If metastasis found, then enucleation is NOT appropriate.
- Much of the treatment protocols were developed from the results of the COMS trial –a randomized prospective treatment study of choroidal melanoma.
- Methods of treatment depend on
- Size, location, extent
- vision of affected + fellow eye
- age + general health
- Treatment Protocols:
- Observation:
- small melanoma <1mm thick
- Elderly or otherwise short life expectancy
- Enucleation: Gold-standard for treatment
- Does NOT disseminate tumor secondary to manipulation.
- Appropriate for all medium to extra-large tumors
- Brachytherapy: most common method of treating uveal melanoma.
- High dose, localized radiation. Usually I125, Ruthenium106 plaques
- Local control rates >96%
- Optic neuropathy + radiation retinopathy in 50%-Location + dose dependent complications
- Observation:
- Charged particle radiation: proton beam.
- Requires tantalum localization clips.
- Up to 98% local control. Increased radiation dose to anterior segment.
- External Beam Radiation: can be used in conjunction with enucleation or brachytherapy.
- Other Alternative Treatments:
- Photoablation + hyperthermia → subretinal fluid treated with grid laser: leads to increase in tumor growth due to rupture of Bruch’s membrane
- Transpupillary Thermotherapy –associated with decrease in tumor volume.
- Cryotherapy –not thought to be effective
- Transcleral diathermy –contraindicated–provides route for extrascleral extension
- Surgical excision: may have success, but difficult + margins are very important.
- Chemotherapy: Not effective for treatment, but may be palliative
- Immunotherapy: under investigation
- Exenteration: rarely employed, but may be used in cases with extrascleral extension.
- Prognosis: 5yr mortality:
- 50% for large
- 30% for medium
- 12% for small
- Risk factors for mortality:
- Large tumor
- Growth
- Anterior tumor location
- Extraocular extension
- Older age
- Tumor regrowth after ocular conservation therapy
- Rapid decrease in size after globe conserving treatment
- Juxtapapillary tumors.
- Histopathological Features associated with increased mortality
- Epithelioid cells
- High mitotic index
- Complex microvascular patterns
- Large nuclei
- Tumor infiltrating lymphocytes
- Monosomy 3
- Trisomy 8
- Collaborative Ocular Melanoma Study.
- 1003 patients with large or extra-large choroidal melanomas
- Greater than 16mm diameterand/or greater than 10mm thick
- Comparedenucleation to enucleation + external beam
- 5-yr survival 57%with enucleation versus 62% with external beam
- No improvement with radiation over enucleation alone.
- 1317 patients with medium-sized choroidal melanoma
- Tumor size: 6mm-15mm diameter and/or 2.5-10mm thick. Compared enucleation with brachytherapy
- No difference in mortality (18% for enucleation and 19%for brachytherapy)
- 43% had visual acuity worse than 20/200 with brachytherapy
- Small ↑ in mortality with tumor recurrence
- 204 patients with small choroidal melanoma
- 4-8mm wide x 1-2.5mm thick
- Mortality 1% at 5 years
- COMS Study risk factors for growth
- Increased thickness + diameter @ presentation
- Orange pigment
- No drusen or RPE changes
- Pinpoint hyperflourescence on FA
- 1003 patients with large or extra-large choroidal melanomas
Pigmented Epithelial Tumors of Uvea + Retina:
- Adenoma:
- (benign) very rare
- Oval, deeply melanotic tumors arising abruptly from RPE
- Rarely undergo malignancy change.
- Adenocarcinoma:
- very, very rare
- Malignantpotential very low.
- Ciliary Cysts
- Opacified ciliary cysts associated with multiple myeloma and macroglobulinemia
Fuchs’ Adenoma

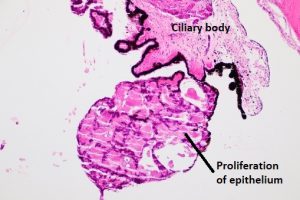
17.22 Fuch’s adenoma
-
- Usually incidental finding @ autopsy.
- Glistening white irregular tumor from ciliary crest.
- Benign proliferation of non-pigmented epithelium
- Acquired Hyperplasia: RPE + ciliary pigmented epithelium may
- Proliferate in response to trauma or surgery.
- may mimic choroidal melanoma
- Treatmentis to observe.
- Combined Hamartoma:
- Rare–occurs at the disc margin. Dark pigmented, minimally invasive lesion
- Proliferation of RPE, glial, + blood vessels
- May cause retinal traction.
- May have more or less pigment
- May be mistaken for melanoma.
Angiomatous Tumors
Choroidal Hemangioma
- Circumscribed choroidal hemangiomas are not associated with systemic disorders
- Red/orange tumor, often in macula
- Often produces secondary retinal detachment
- Often affect overlying RPE with degeneration of outer retina.
- Differential diagnos is
- Amelanotic choroidal melanoma
- Choroidal osteoma
- Metastatic carcinoma
- Granuloma of choroid.
Diffuse choroidal Hemangioma associated with Sturge –Weber Syndrome
- Diffuse reddish-orange thickening of retina
- Tomato catsup fundus.
- Retinal detachmentcommon
- Glaucoma common.
- FA shows large choroidal vessels, in pre arterial + arterial phases and late staining of tumor. NOT pathognomonic. U/S shows high internal reflectivity, acoustic heterogeneity. No shadowing. CT scan can be helpful in differentiating from osteoma. Treatment:
- observation if asymptomatic
- Serous detachment (including fovea) → photocoagulation
- Treating withlight laser to adhere retina + ↓ serous fluid.
- Recurrent detachments common.
- PDT → to entire lesion may involute tumor + ↓ fluid.
- Brachytherapy, Proton Beam, Gamma Knife have been used to involute tumor as well.
- Periocular + intraocular avastin is being investigated.
Retinal Angiomas:
Capillary Hemangioma
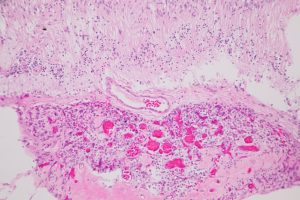

17.23 Capillary hemangioma in Von Hippel –Lindau syndrome
- Rare, autosomal dominant (1 in 40,000)
- Diagnosis at age 10-30.
- Orange/red tumor arising in retina with tortuous vessels.
- Subretinal exudates common + often in fovea.
- Exudate detachment common.
- “Von Hippel disease”
- familial in 20%
- Bilateral in 50%
- Von Hippel–Lindau Syndrome choroidal hemangioma with associated with cerebellar hemangioblastoma
- Genetics
- Gene located on chromosome 3
- Associated tumors
- Retinal capillary hemangioma
- Cerebellar hemangioblastoma
- Renal Cell Carcinoma
- Pheochromocytoma
- Genetic screening can help determine risk of tumors,
- FA shows feeding arteriole, massive network + draining venule.
- Treatment:
- Photocoagulatesmall lesions.
- Cryotherapy for larger, peripheral lesions.
- Penetrating diathermy for very large lesions.
- Avastin isbeing investigated.
Cavernous Hemangioma


17.24 Cavernous hemangioma
- Uncommon, cluster of grapes lesion.
- Generally not associated with exudates.
- Small hemorrhages + gliosis may appear on surface.
- May occur on optic disc.
- FA is diagnostic–fills slowly fluorescein + RBCs separate.
- No vitreous leakage (unlike Coats Disease or capillary hemangioma)
- No treatment necessary for cavernous hemangioma.
- Composed of dilated, thin walled vessels, interconnected
A-V malformation
A-V malformation:
- Racemose Hemangioma (congenital retinal A-V malformation)
- Range from small vascular communication near disc or periphery
- To large tangle of tortuous vessels throughout fundus.
Wyburn-Mason:
- Racemose hemangioma + midbrain A-V malformation.
- Associated A-V malformations possible in orbit+ mandible
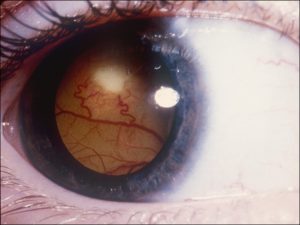
Retinoblastoma

17.25 Retinoblastoma
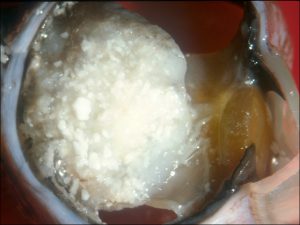
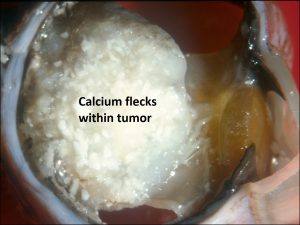
17.26 Gross cross-section globe of RB
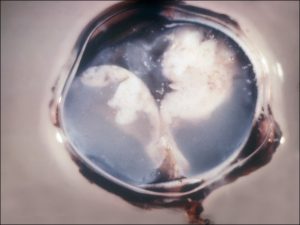
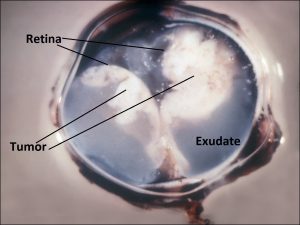
17.27 Exophytic growth pattern of RB
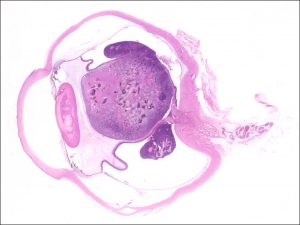
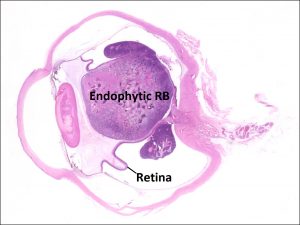
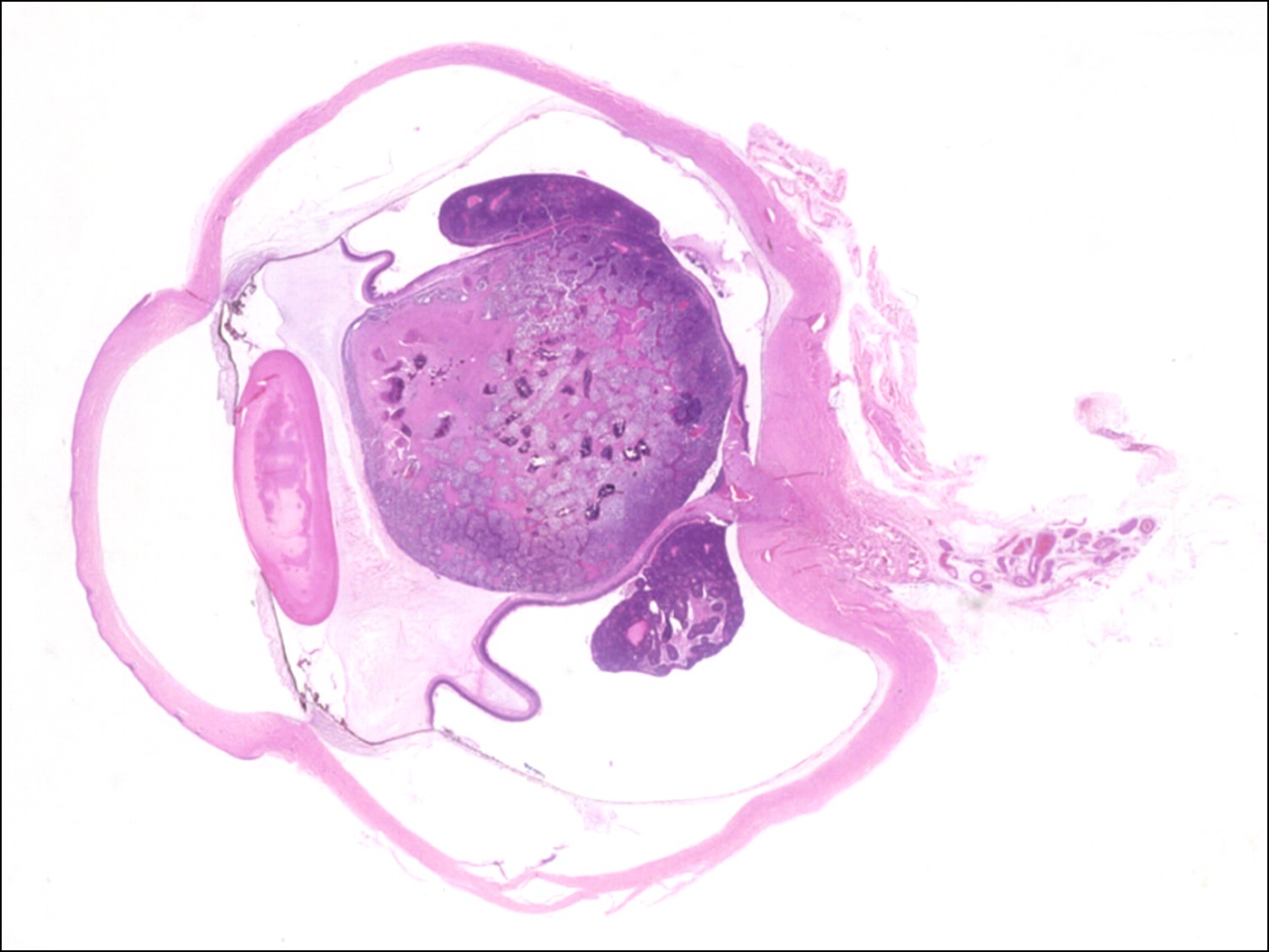
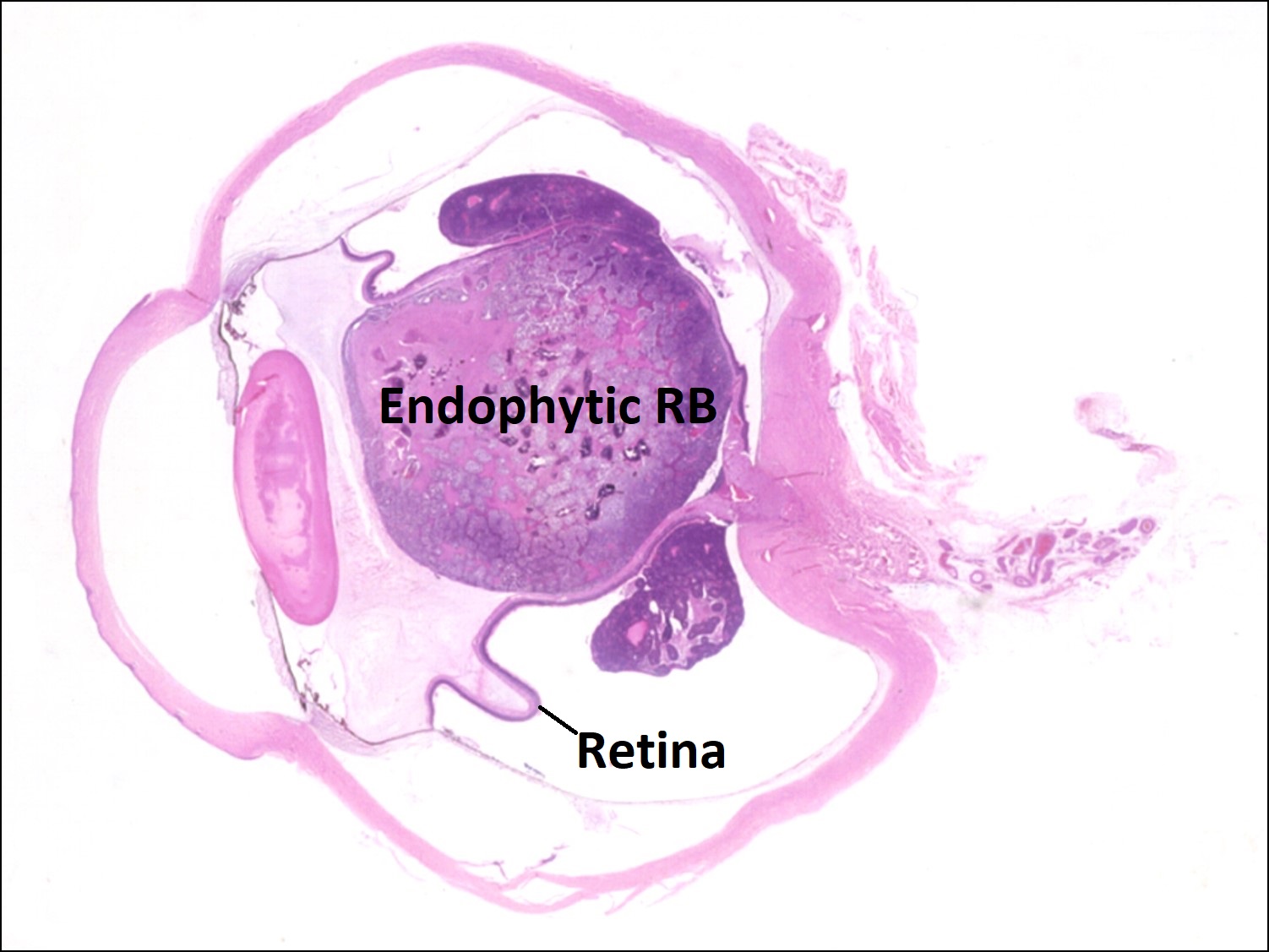
17.28 Endophytic growth pattern

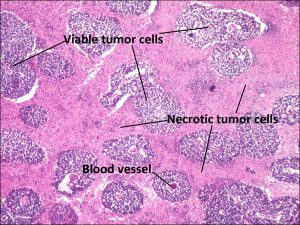
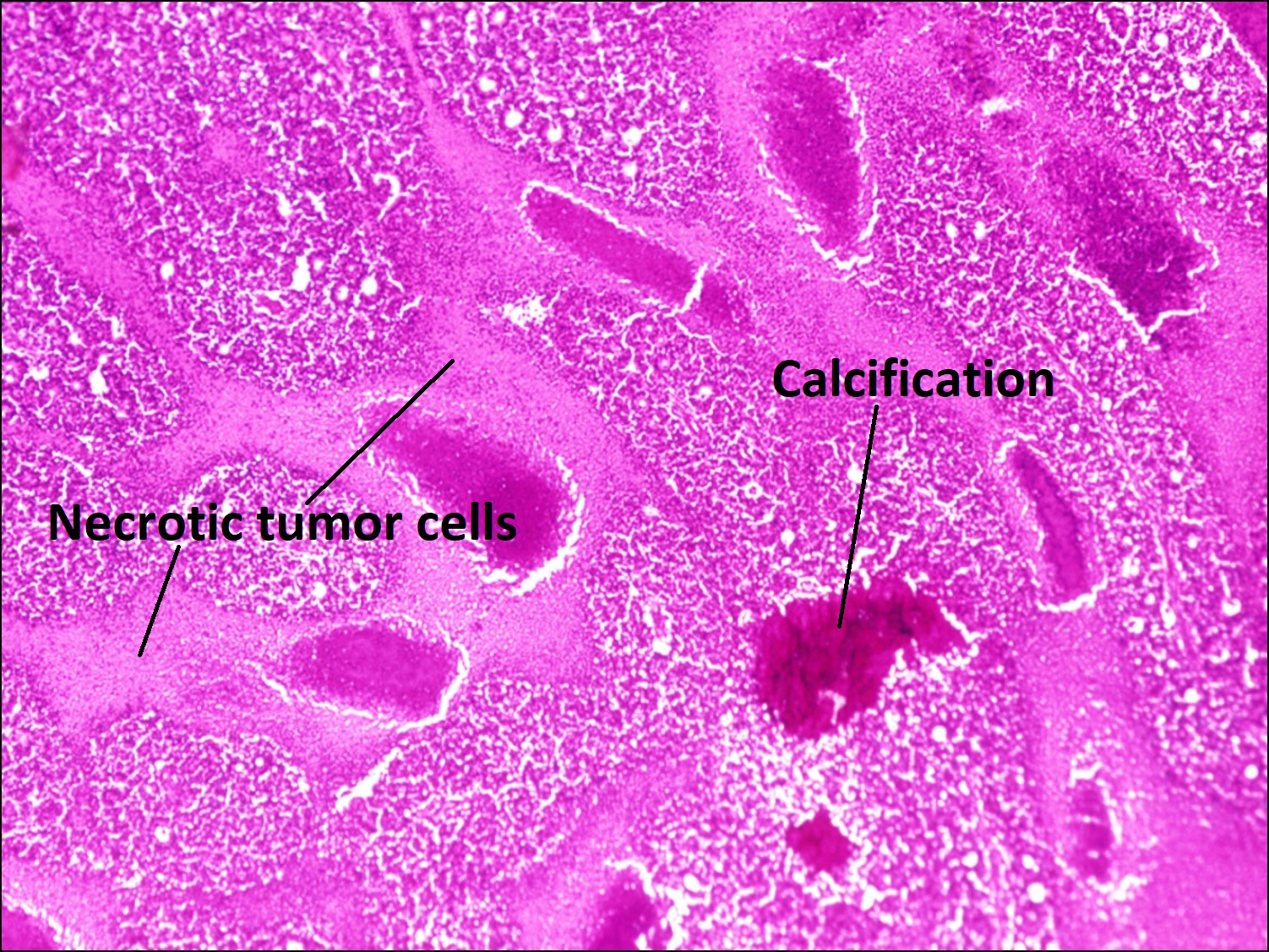
17.29 Necrosis
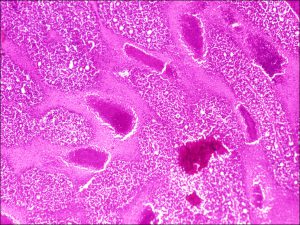
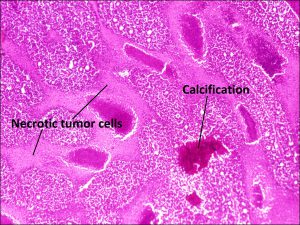
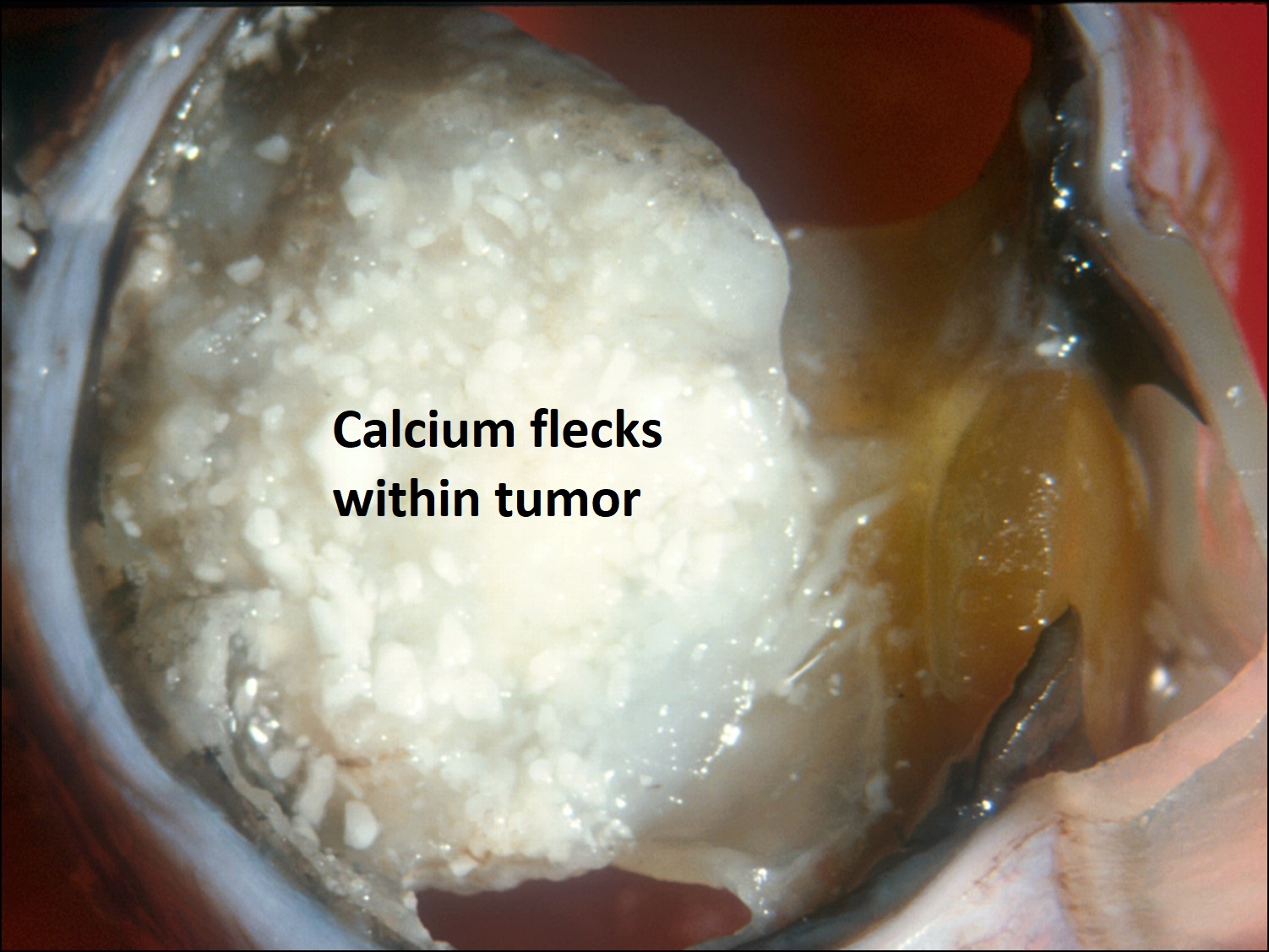
17.30 Dystrophic calcification

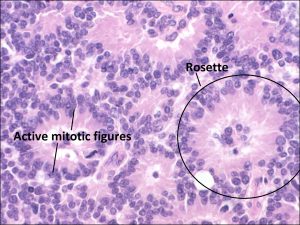
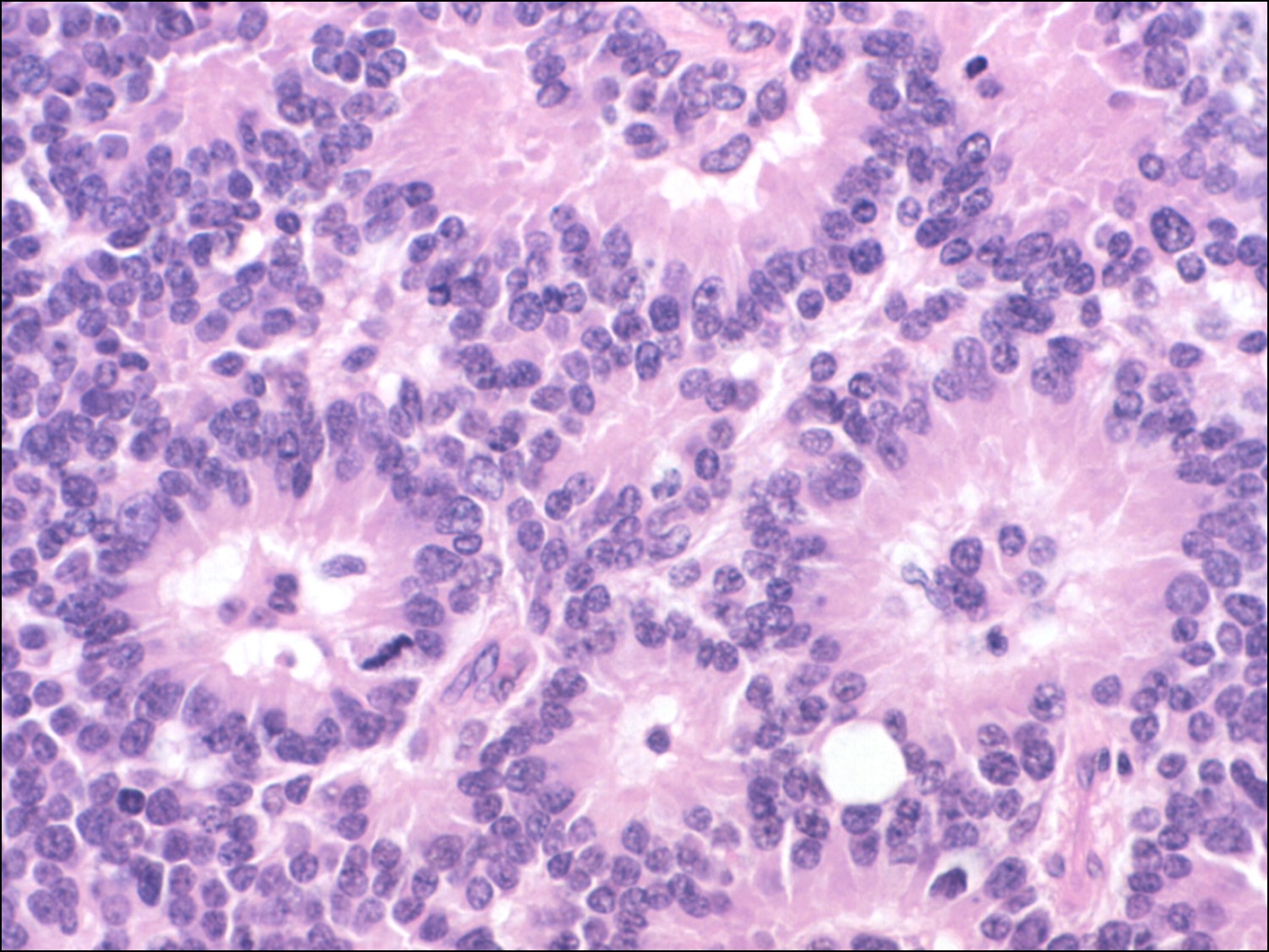
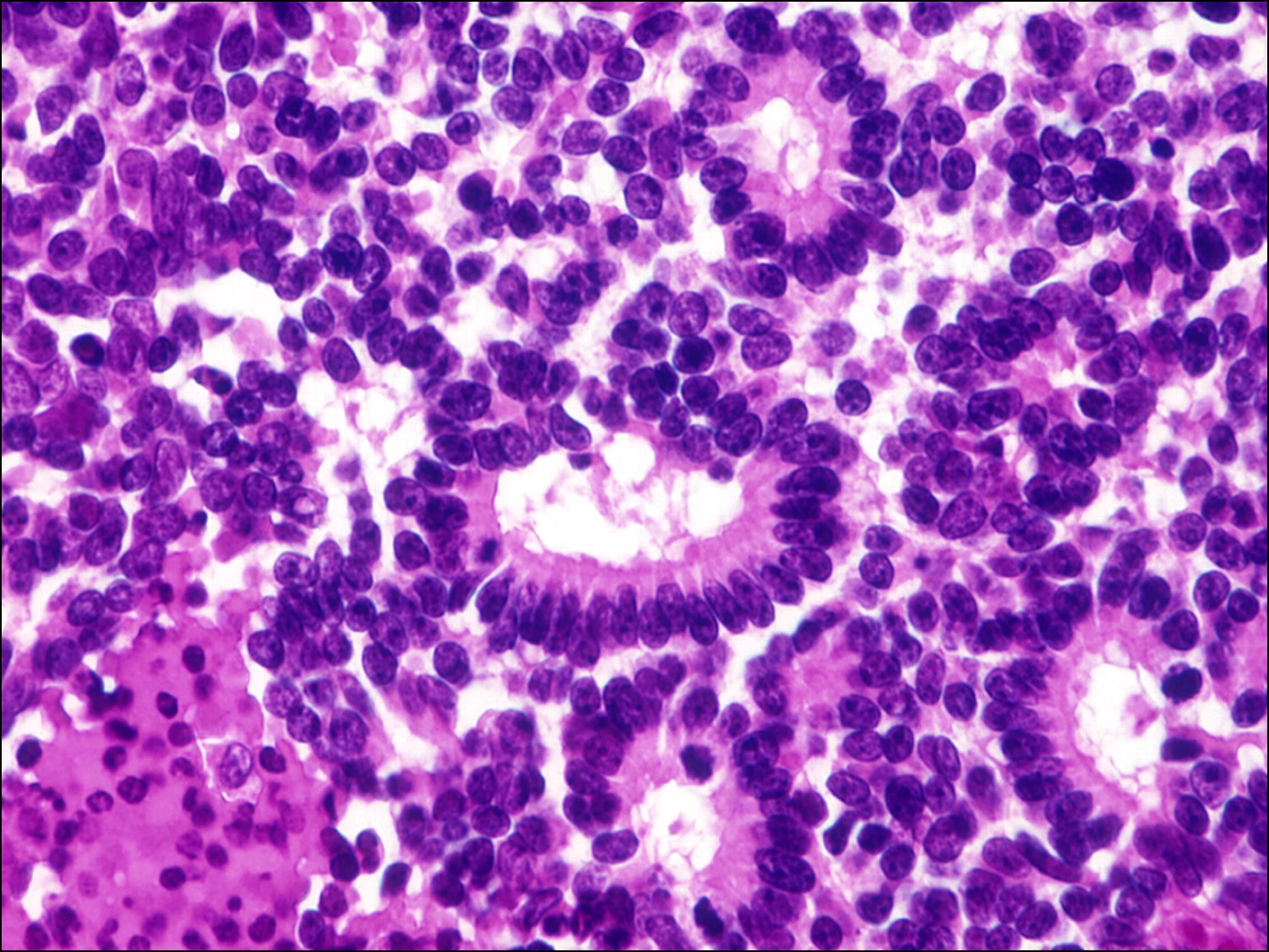
17.31 Flexner-Wintersteiner rosettes
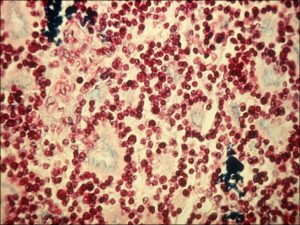
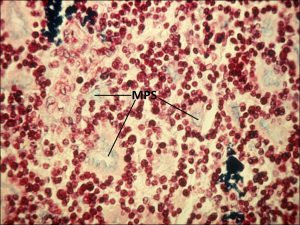
17.32 Alcian blue stain


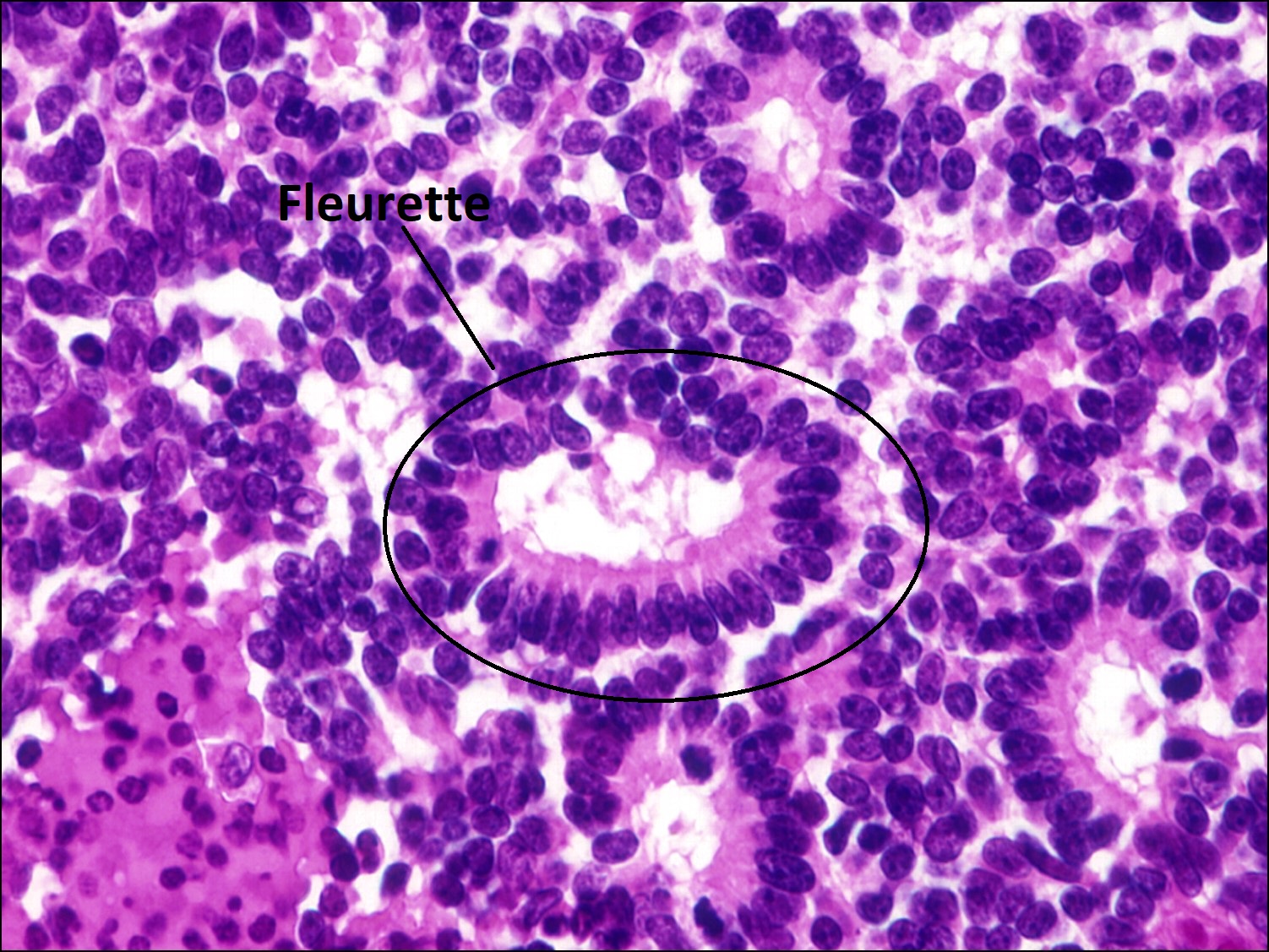
17.33 Fleurette

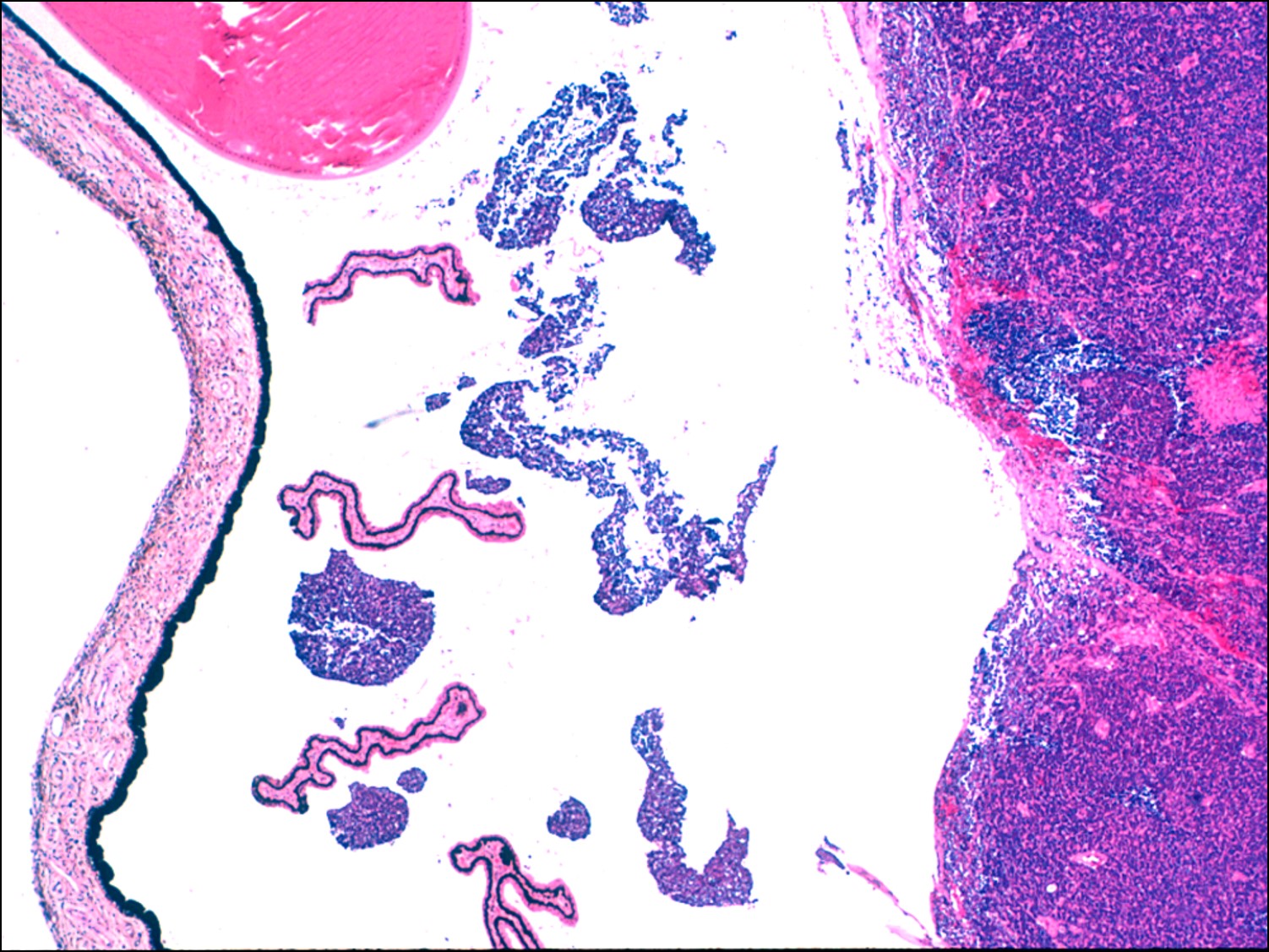
17.34 Vitreous seeding

17.35 Tumor invading optic nerve
Nick’s Tips: The differential diagnosis of leukocoria in a child and specifically the details of retinoblastoma is one of the most high yield study subject for the Boards. In addition, this material may enable you to save a child life someday.
Retinoblastoma:
- #1 most common primary malignancy of childhood.
- 2nd most common primary malignancy (behind uveal melanoma) in all age groups.
- 1 in 14,000-20,000, 300 new cases/year in USA.
- No gender predilection
- 30% to40%bilateral.
- Age @ diagnosis:
- Family history of known retinoblastoma→ 4months
- Bilateral disease → 14 months
- Unilateral disease →24 months
- 90% by age 3.
- Genetics: mutation in retinoblastoma tumor suppressor gene.
- 6% familial
- 98% chance of germline mutation in bilateral disease.
- Children of retinoblastoma survivor have 45% chance of having retinoblastoma (50% transmission, 90% penetrance)
- 94% sporadic.
- About 60% have unilateral disease
- No germline mutation
- Most have unifocal tumor
- 40% have new germline mutation + multifocal disease.
- About 60% have unilateral disease
- About 95% chance of finding germline mutation with genetic testing.
- 6% familial
- Presentation:
- Leukocoria = most common presentation
- Strabismus + ocular inflammation also possible.
- Clinical Appearance
- Begins as grey/translucent intraretinal tumor with dilated feeder vessels
- Becomes chalky white as calcification occurs.
- Serous retinal detachment may occur as tumor grows + cause ↓ vision + difficulty visualizing tumor.
- Exophytic tumors: grow beneath retina → retinal detachment
- Endophytic tumors:
- Grow on retinal surface into vitreous.
- Vitreous seeding more common
- May seed Anterior chamber leading to
- Pseudohypopyon
- Rubeosis + secondary to glaucoma in 50%
- Diffuse –infiltrating retinoblastoma:
- Appears like intermediate uveitis with diffuse vitreous cells,
- Poor view of retina.
- Metastatic
- Most common metastasis is through direct extension via optic nerve.
- May extend into subarachnoid space.
- May also erode through sclera or emissary veins into orbit.
- In A/C, may invade trabecular meshwork + lymphatics.
- Metastatic to:
- Skull bones, distant bones
- Brain, spinal cord
- Lymph nodes (preauricular/cervical)
- Abdominal viscera
- Clinical Exam:
- Need exam under anesthesia with scleral depression.
- Document clearly location + size of multiple tumors.
- Ultrasound may show calcifications
- MRI is imaging modality of choice for globe, ON, brain.
- Systemic evaluation not necessary if no signs of extraocular extension.
- LP if extensionvia ON thought to have occurred
- Examine siblings + parents
- Differential Diagnosis of Leukocoria:
- Retinoblastoma
- Coats disease
- Astrocytic hamartoma –tuberous sclerosis
- Retinal capillary hemangiomatosis
- Granulomas (especially associated with nematode endophthalmitis) -toxocariasis
- Coats
- Persistent fetal vasculature
- Ocular toxocariasis
- Retinopathy of prematurity
- Retinal detachment
- Coloboma
- Myelinated nerve
- Toxoplasmosis
- Retinal dysplasia
- Trisomy13 → cartilage in ciliary body
- Norrie’s disease
- Retinoschisis
- Cataract (torch, congenital)
- Cyclitic membrane
- Medulloepithelioma
- Incontinenti pigmenti
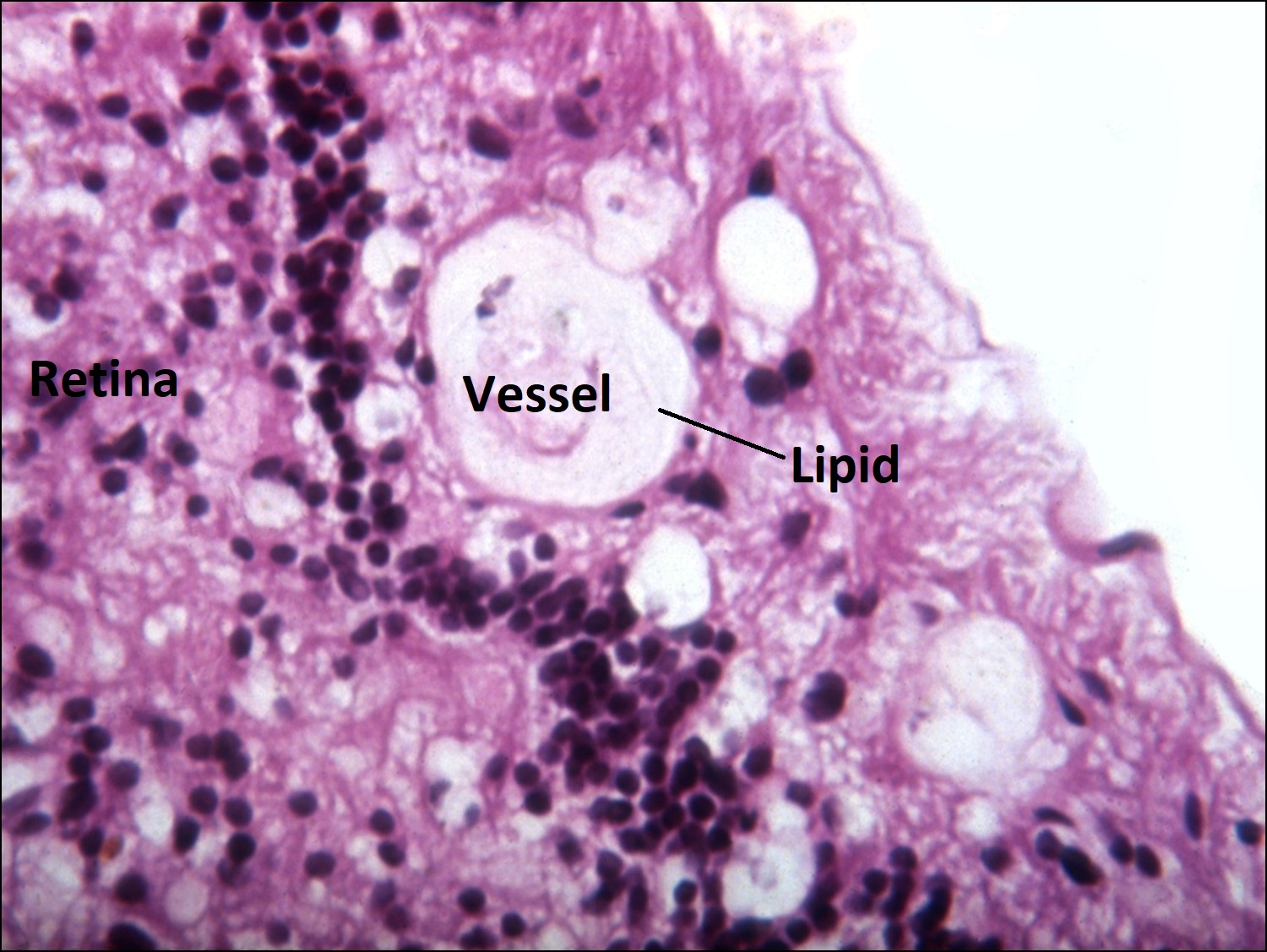
Coats Disease:

17.36 Fundoscopic photo
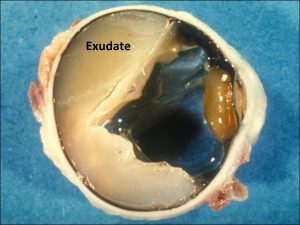
17.37 Gross cross-section globe

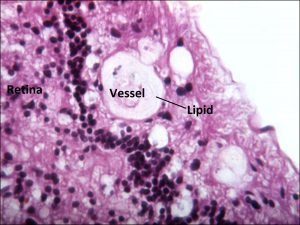
17.38 Telangiectasic vessels leak into retina and under retina
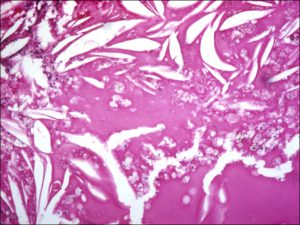
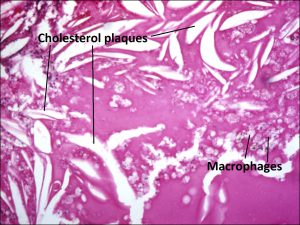
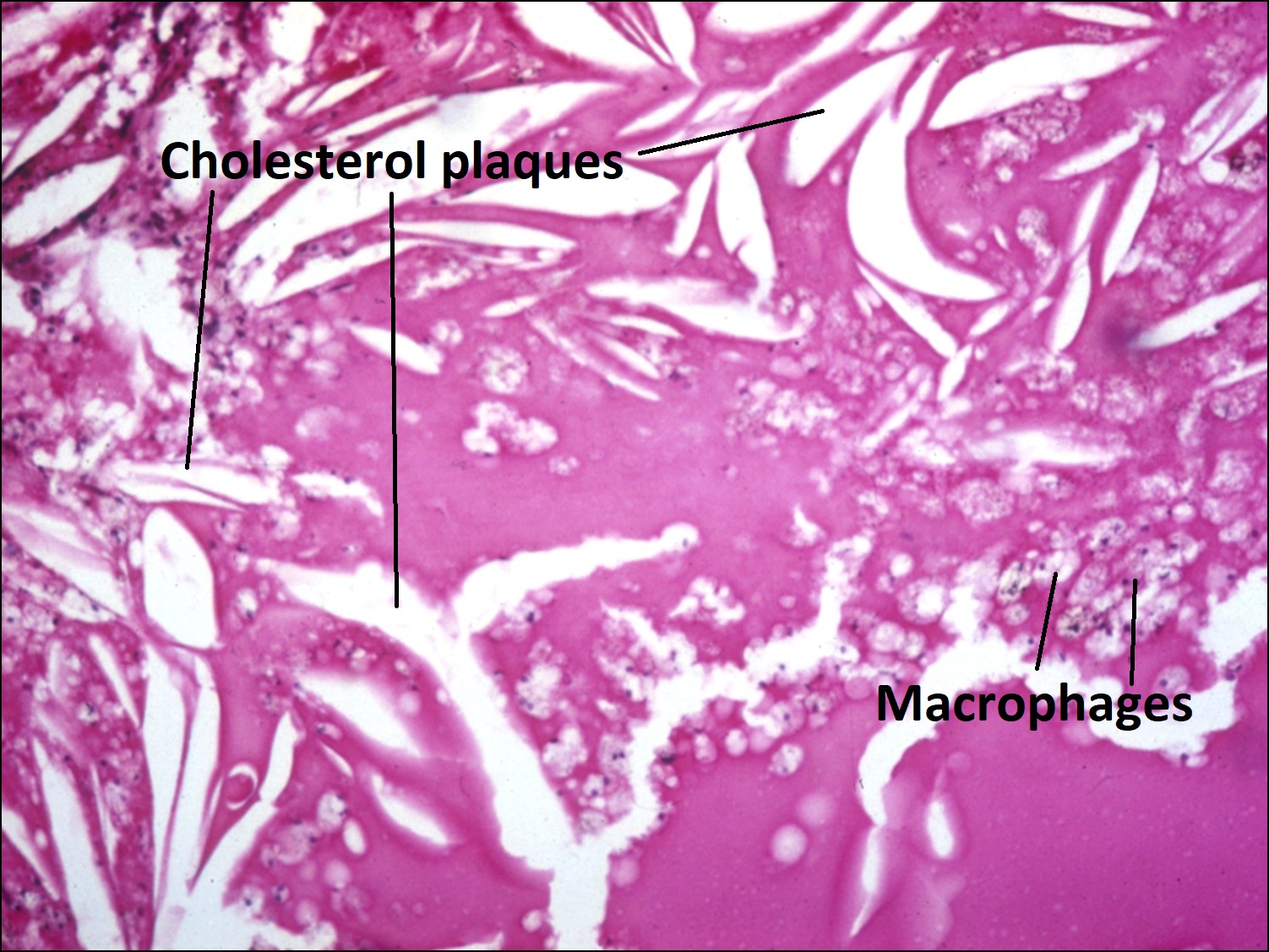
17.39 Processed specimen leaving sickle shaped cholesterol plaques
- male>female (10:1)
- poor visual prognosis
- Unilateral retinal telangiectasias, yellow exudates.
- No distinct mass, (cholesterol rich sickle shaped spaces)
- Fluid leakage may lead to serous retinal detachment which may be large
- Ultrasound documents absence of retinal tumor.
- Treat with laser or cryo of vascular anomalies to decrease serous detachments
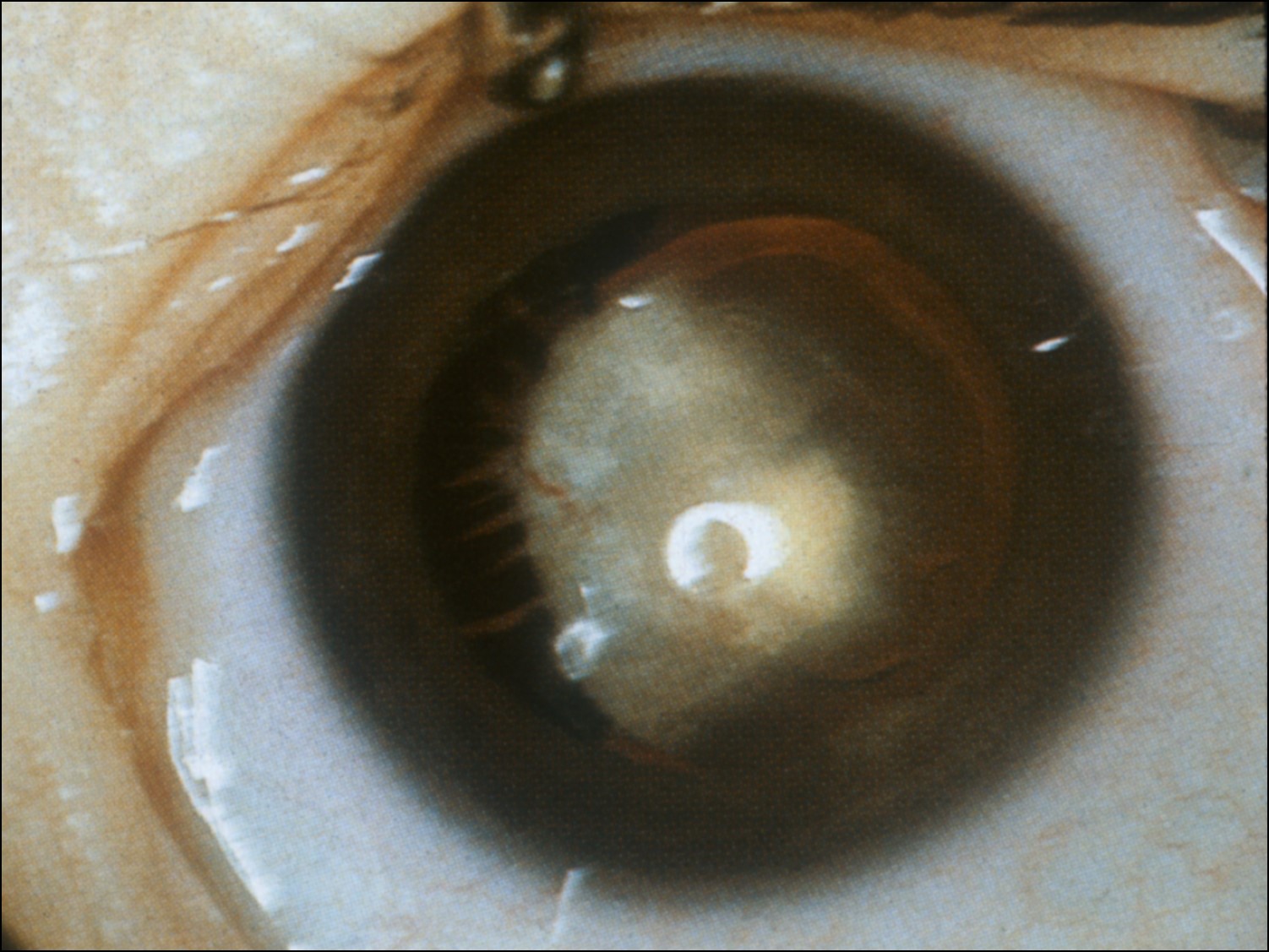
Persistent Fetal Vasculature (Persistent hyperplastic primary vitreous):
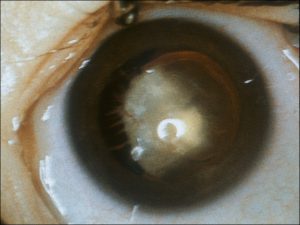
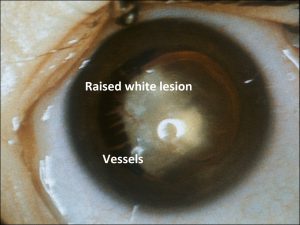
17.40 External slit lamp photo
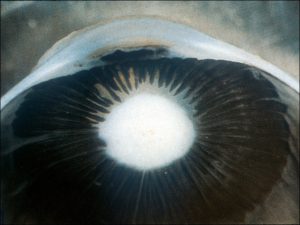
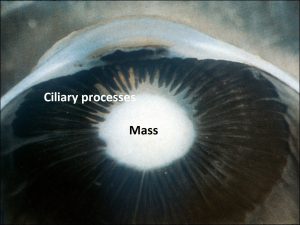
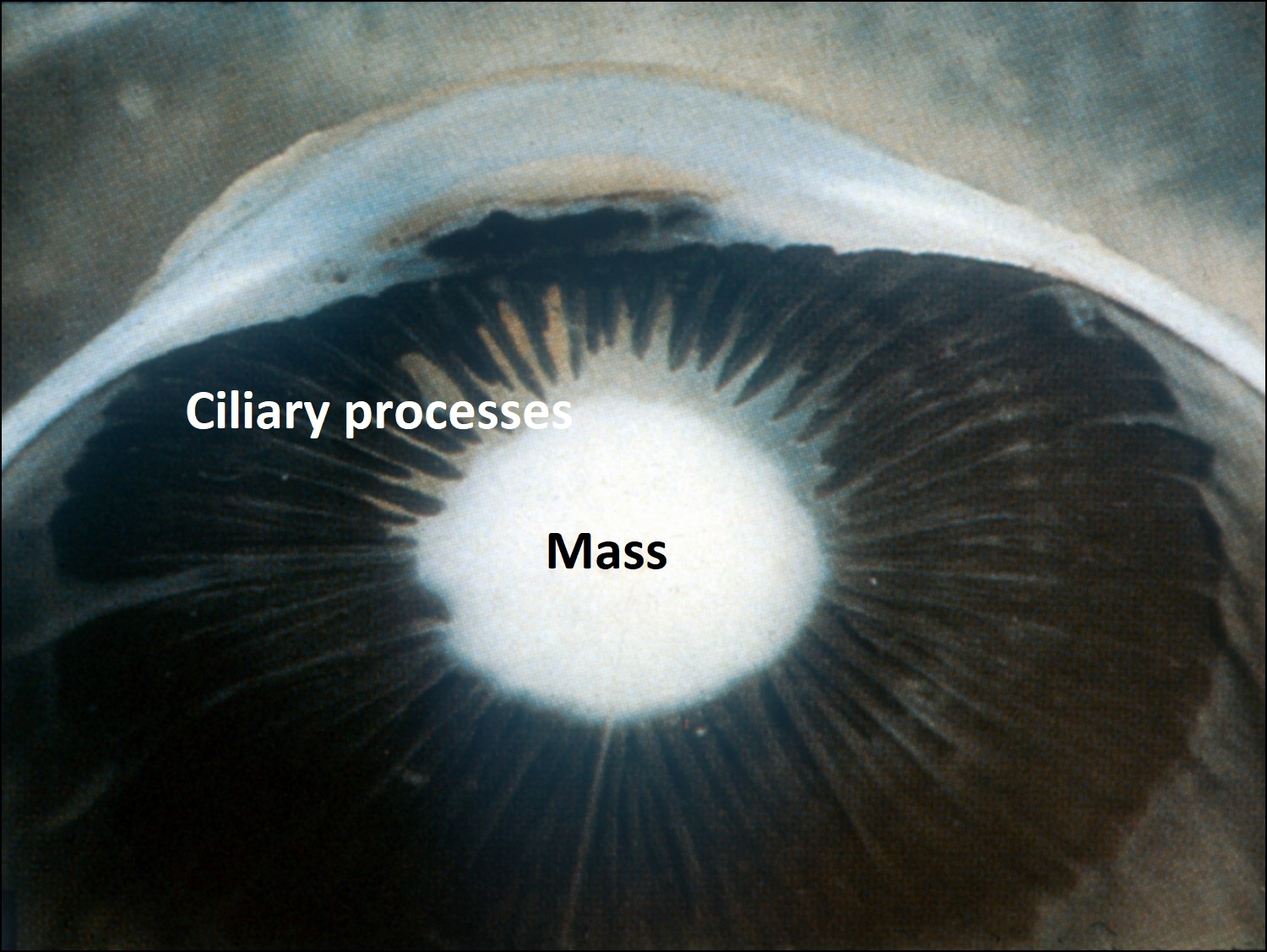
17.41 Mass in the lens pulls and contracts ciliary processes forward.
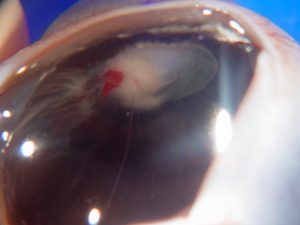
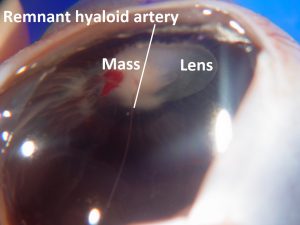
17.42 Remnant hyaloid artery

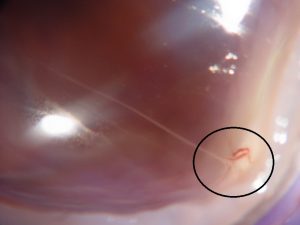
17.43 Bergmeister papillae

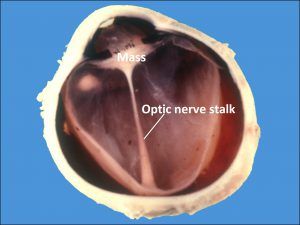
17.44 Gross cross-section glob
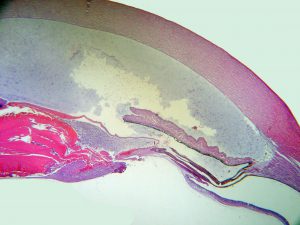
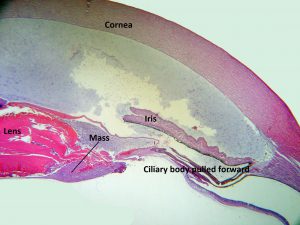
17.45 PHPV
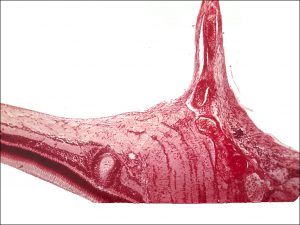

17.46 Remnant Hyaloid artery
- Typically recognized in 1st few weeks of life
- Unilateral in 2/3
- Associated with
- Microphthalmos.
- Shallow anterior chamber
- Hypoplastic iris
- Retrolenticular mass of fibrovascular tissue
- Vascular stalk may be seen arising from nerve.
- May have closed funnel retinal detachment
- Ultrasound shows no tumor, short axial length +/-calcification
- Treatment is vitrectomy/lensectomy.
Ocular Toxocariasis:
- Clinical Evaluation: (yearly follow-up)
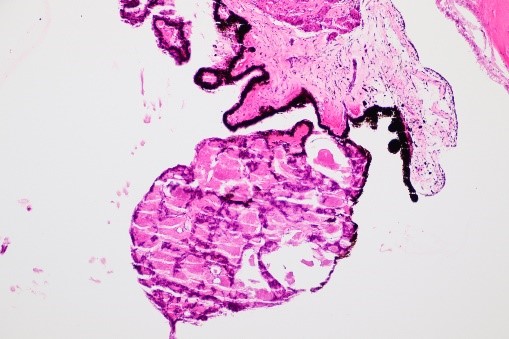

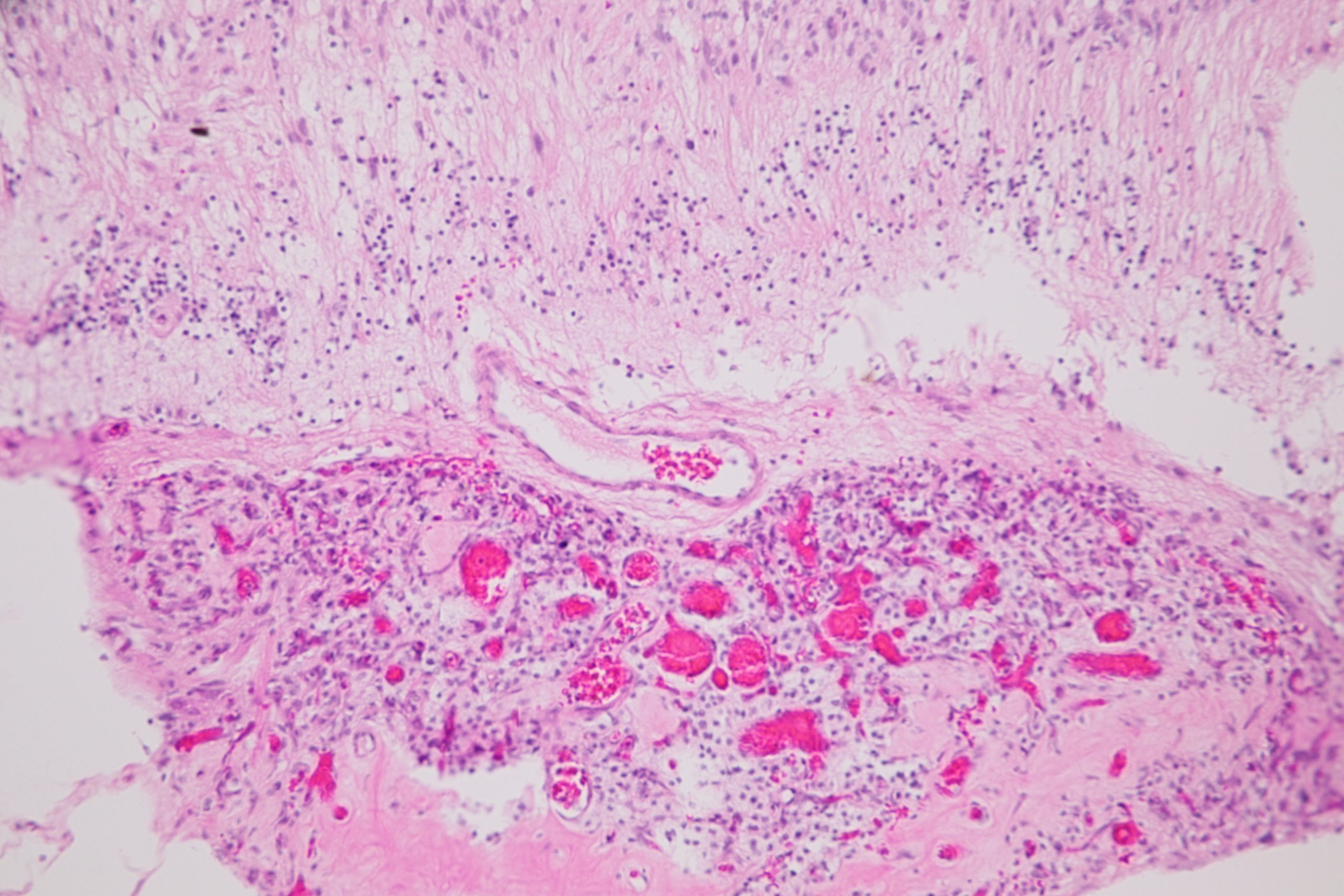
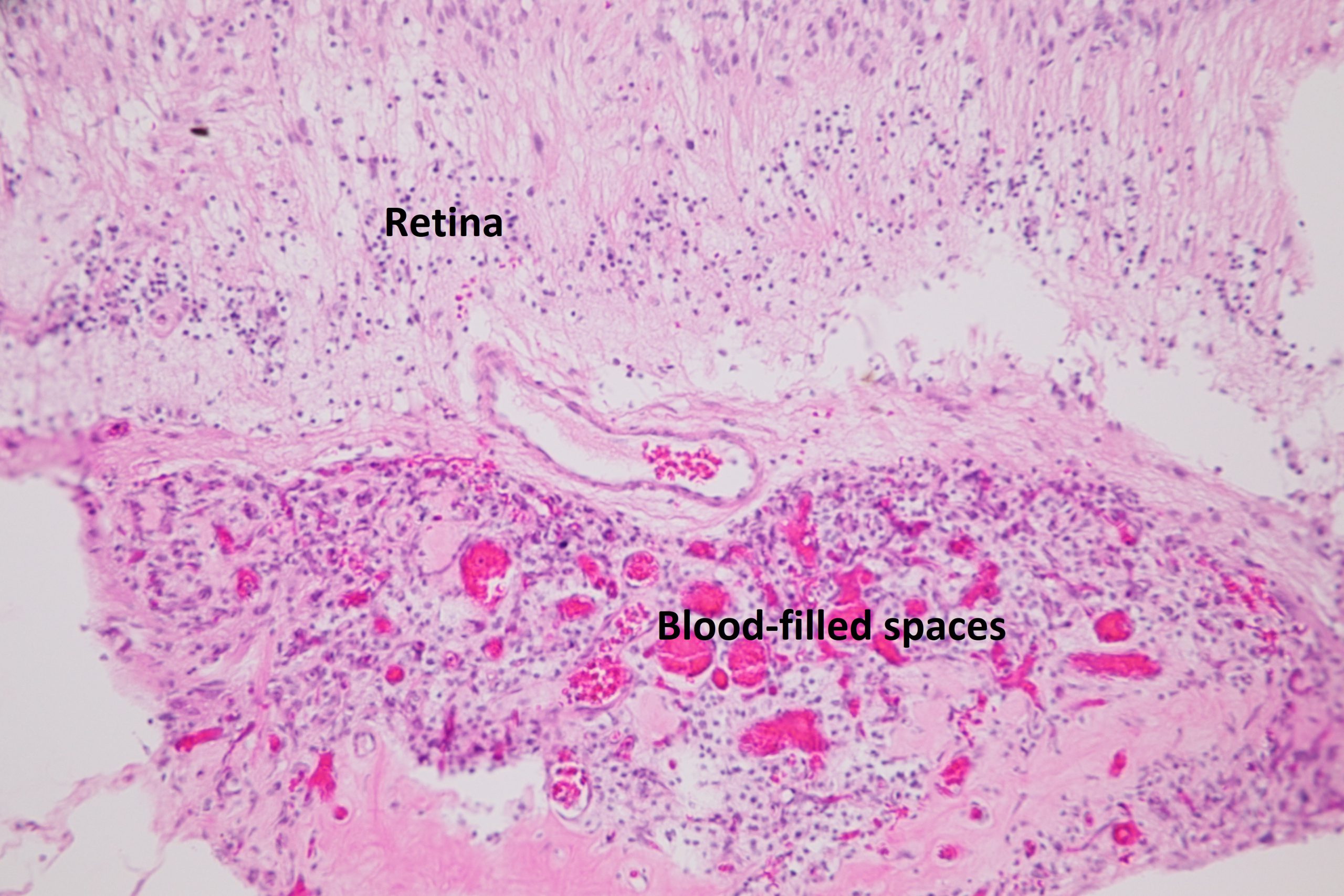

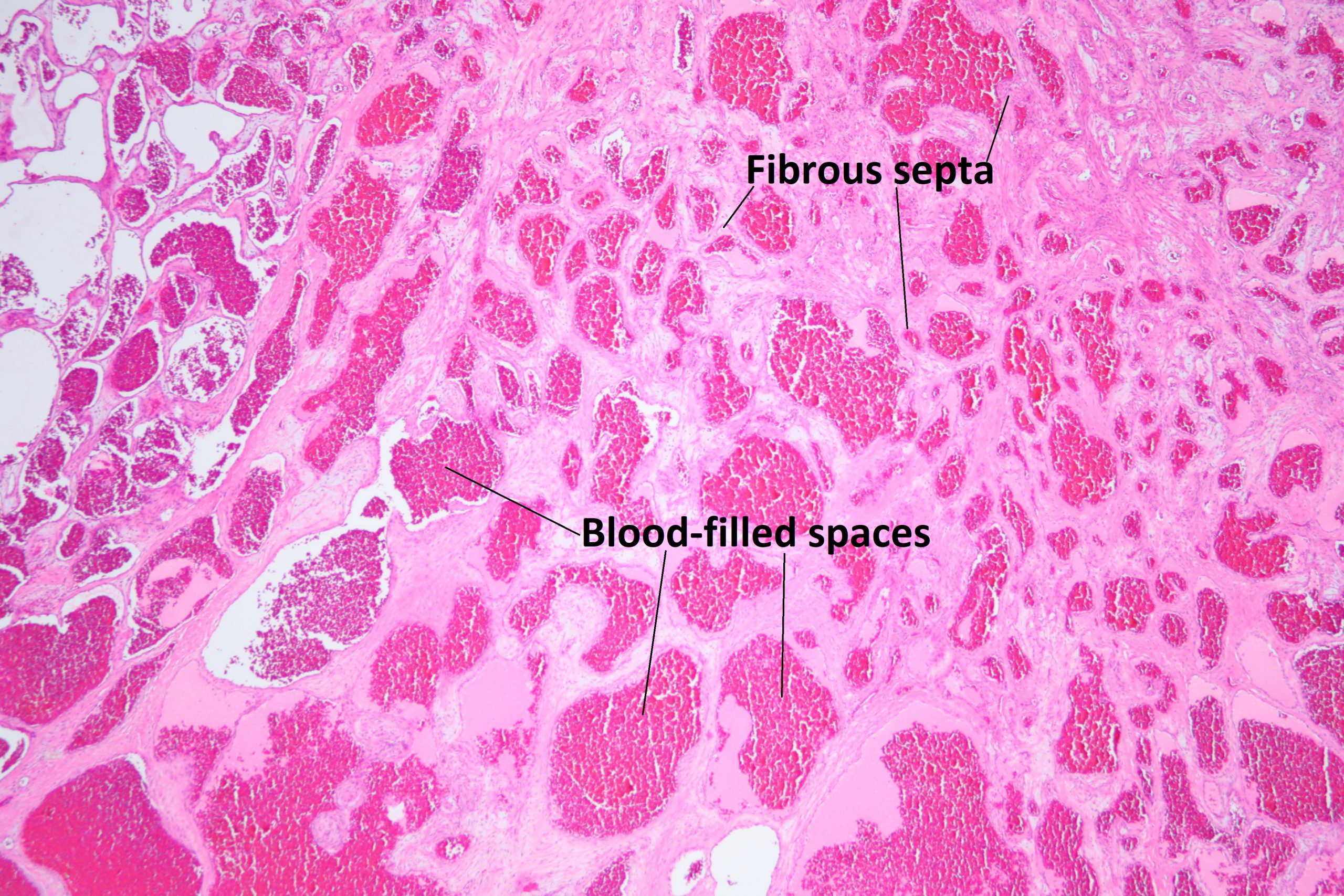
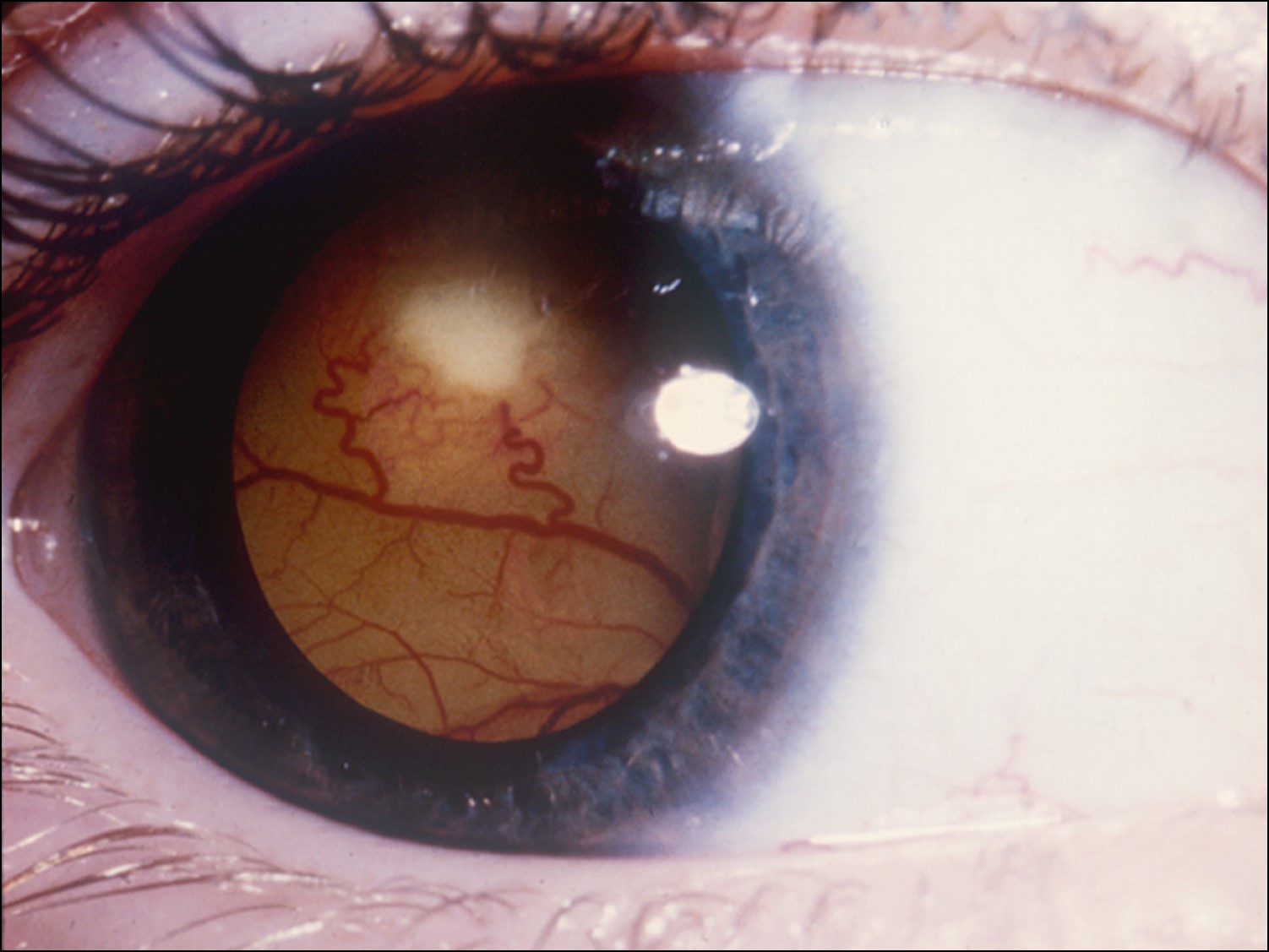











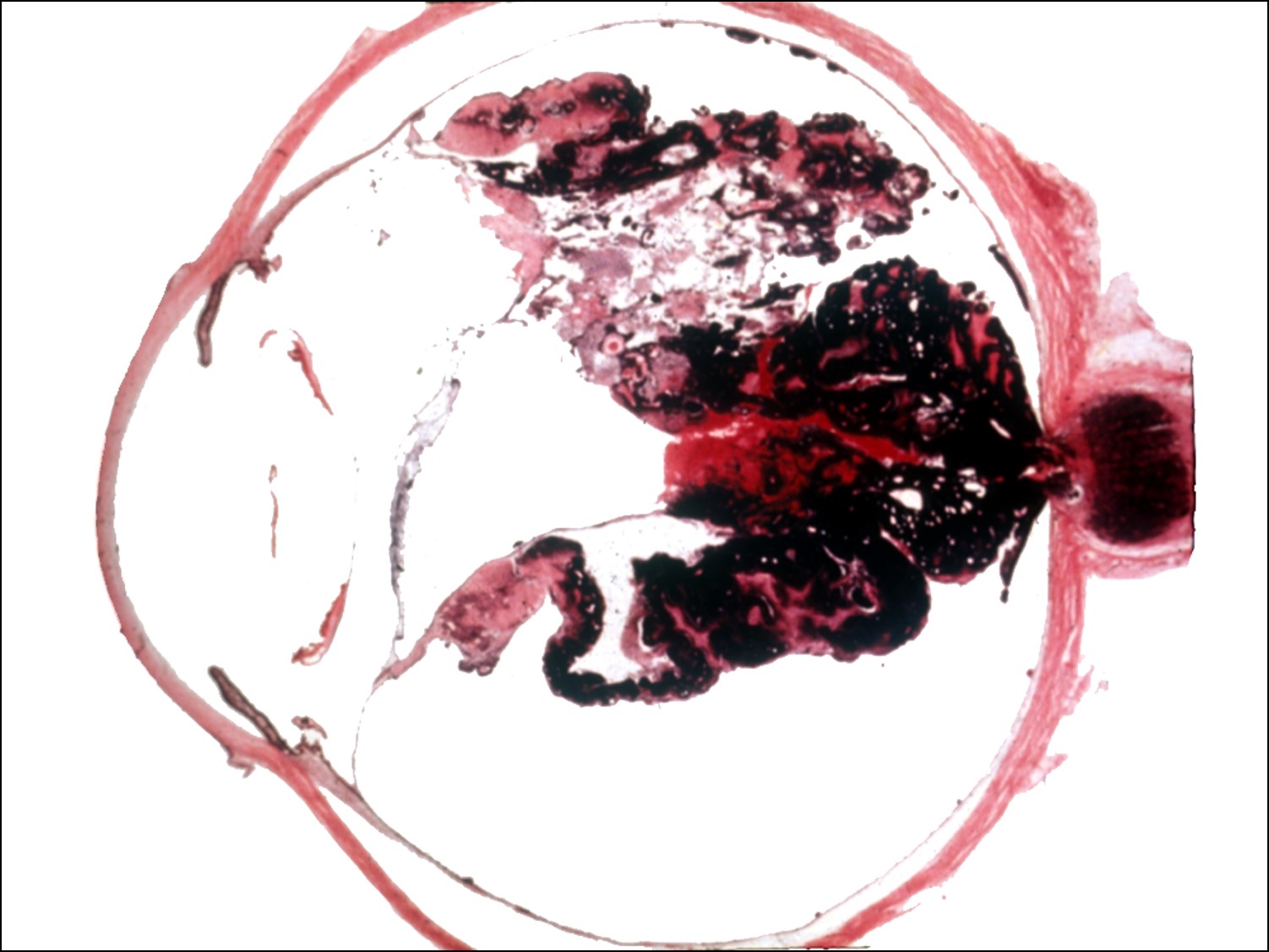
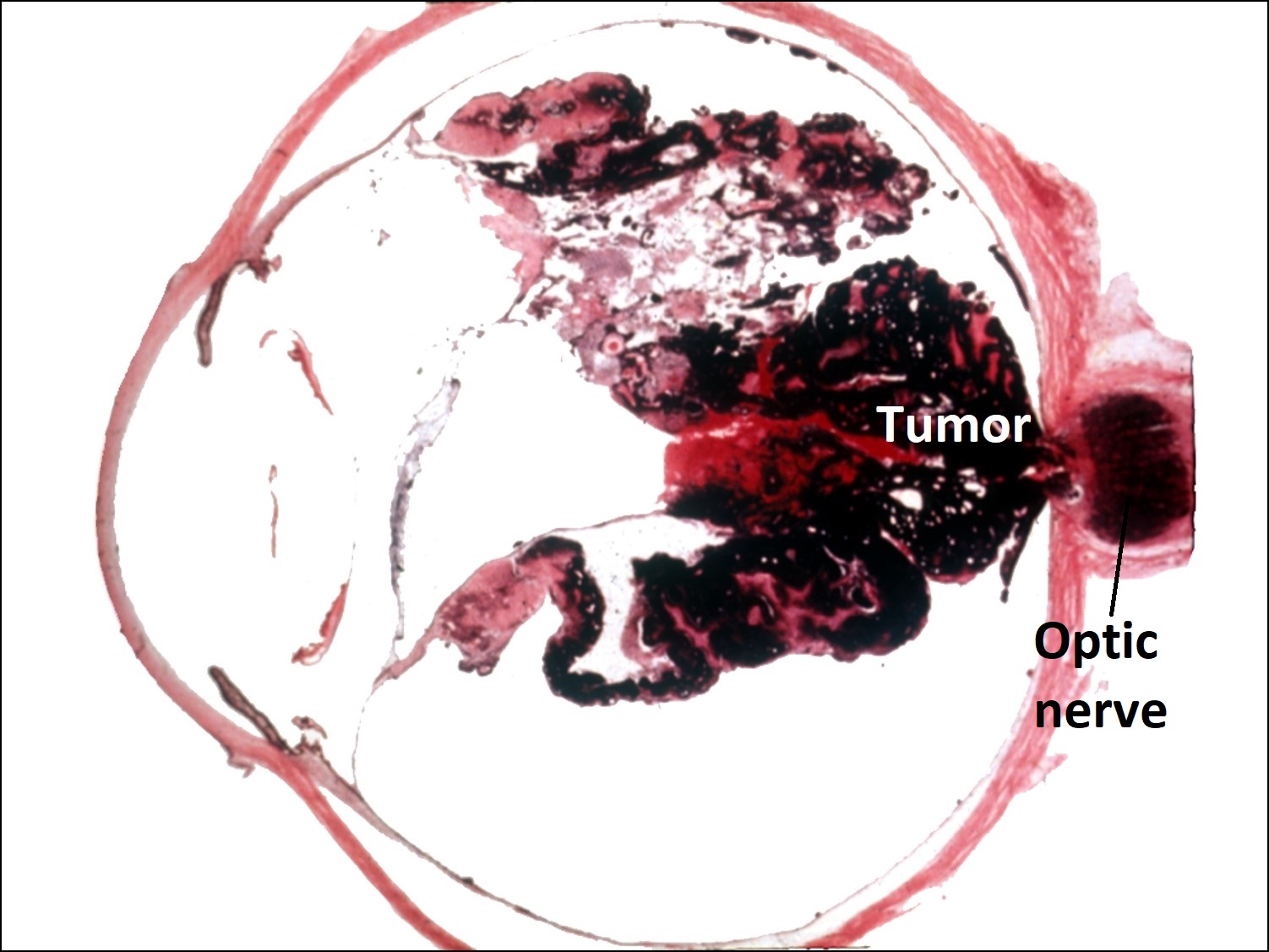


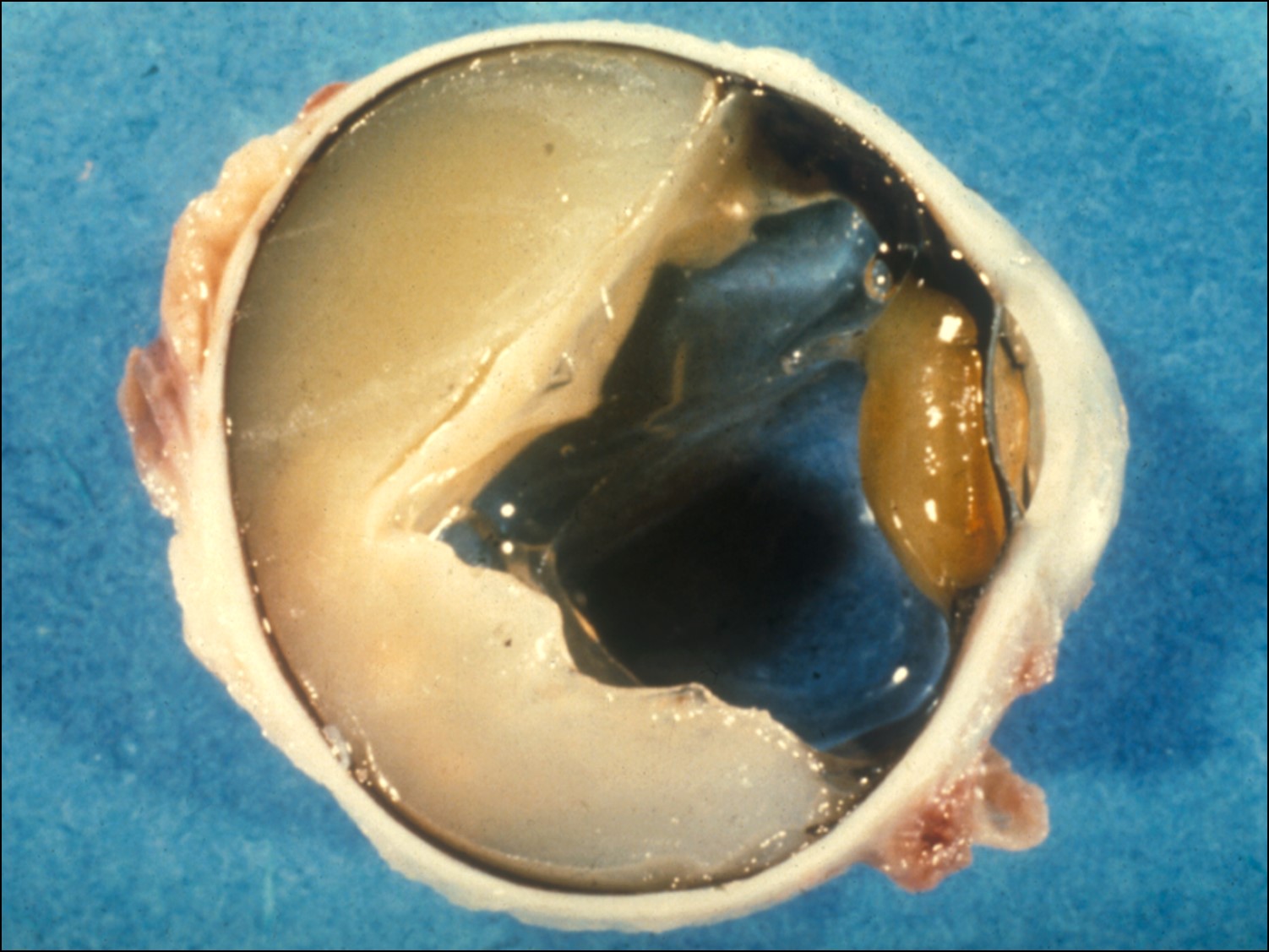
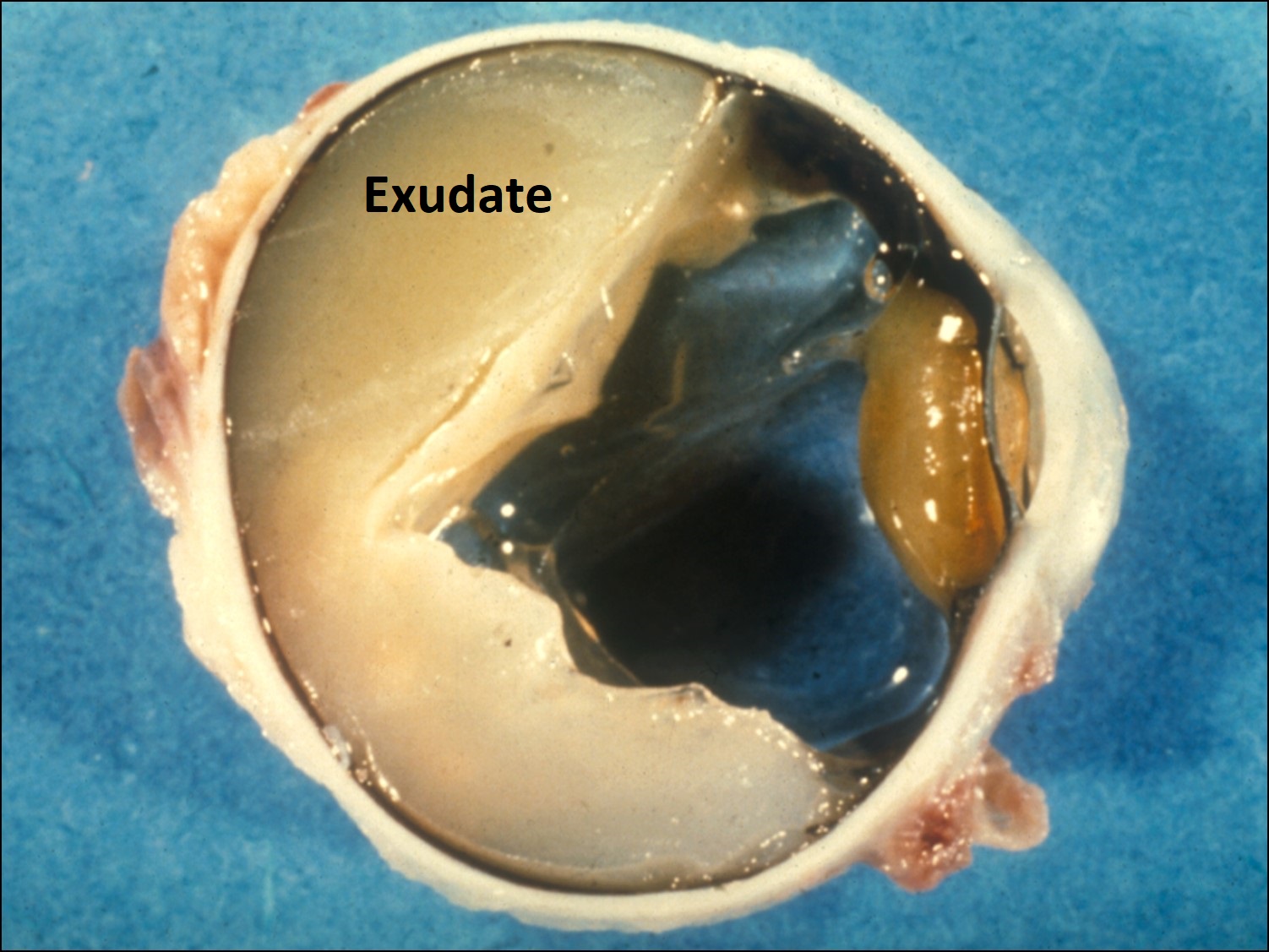




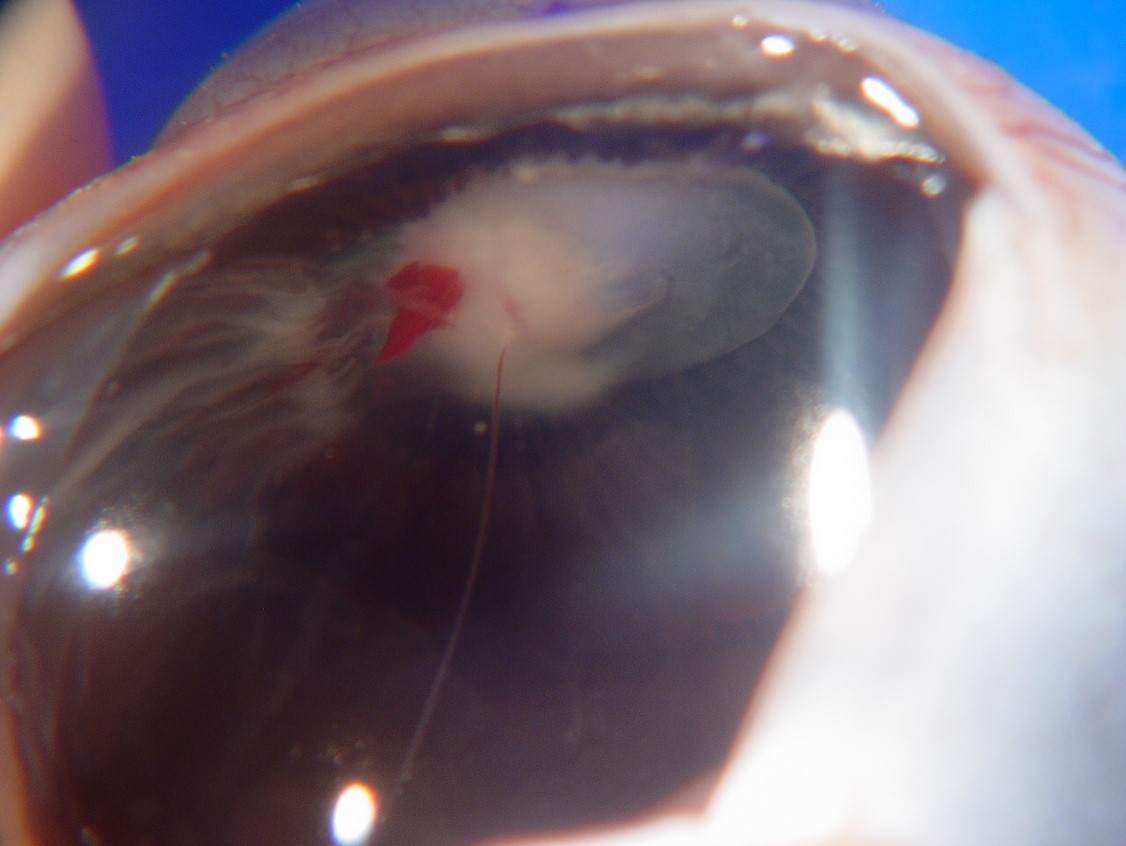


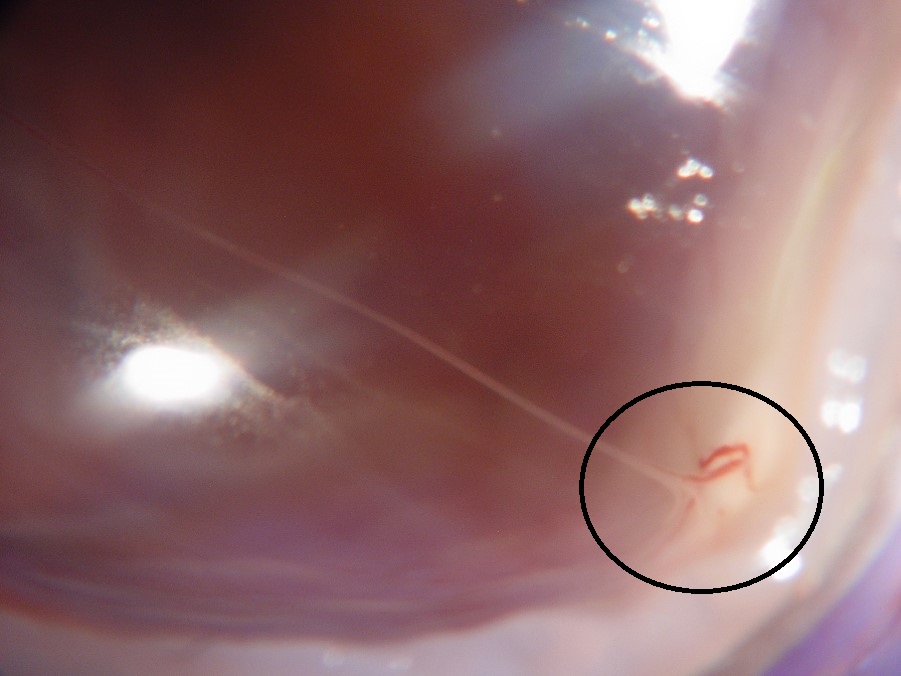

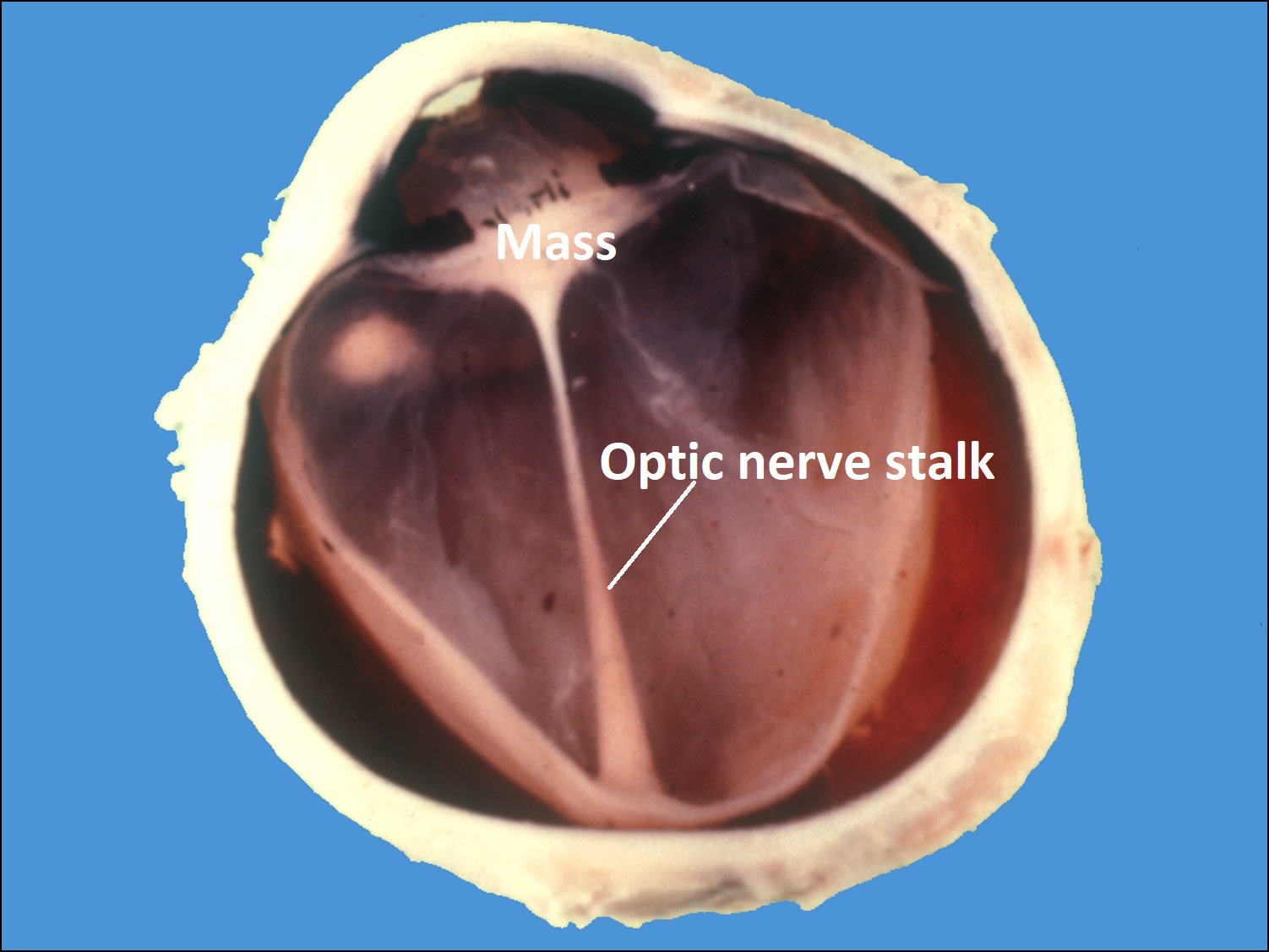


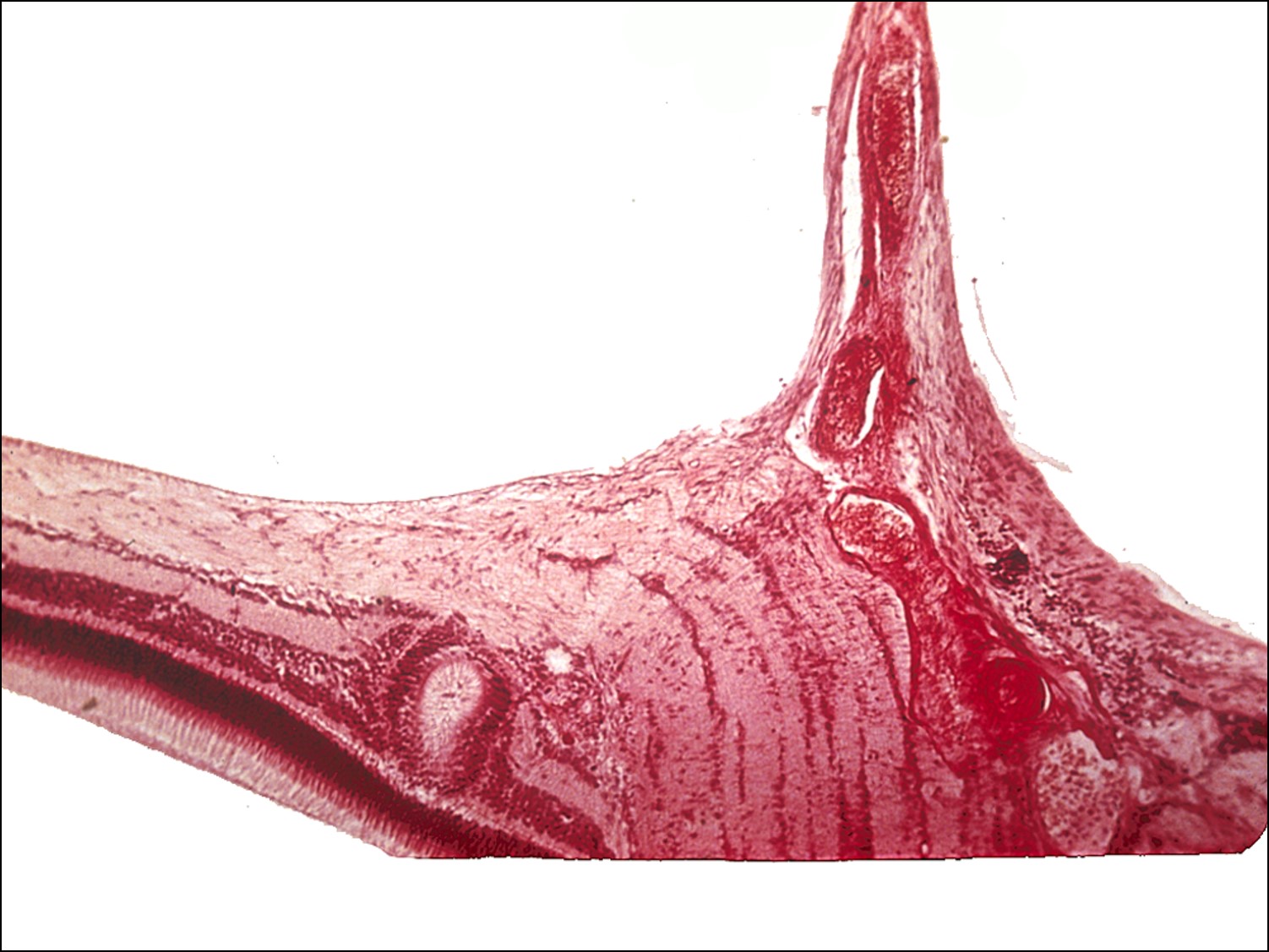
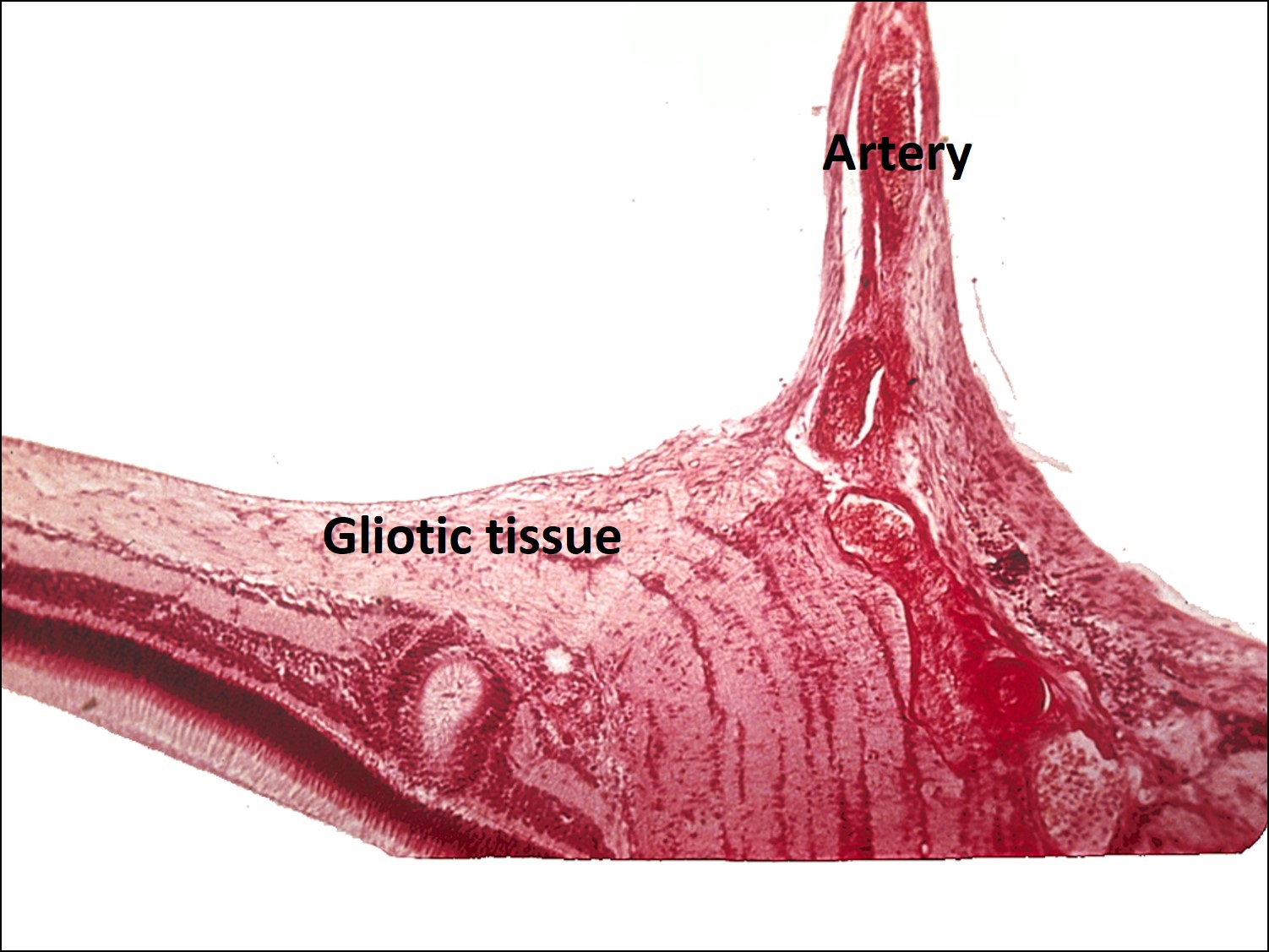


17.47 Toxocariasis
-
- Most patients have a history of soil ingestion or exposure to puppies.
- Posterior mass + peripheral granulomas + uveitis.
- Ultrasound shows vitritis, granuloma, retinal traction, NO calcification
- Astrocytoma: (astrocytic hamartoma)
- Small, white, glistening tumor of nerve fiber layer
- Sometimes large, mulberry appearance with calcification.
- Usually arise from optic disc (termed “giant drusen”)
- Common in tuberous sclerosis + may occur in neurofibromatosis
- Usually ∅assoc. with phakomatosis.
- Retinocytoma: clinically indistinguishable from retinoblastoma
- Histologically benign.
- Same genetic implications as retinoblastoma
- Unclear developmental biology.
- Reece-Ellsworth classification of intraocular retinoblastoma.
- Predicts eye preservation when treated with external beam radiation.
| A | B | |
|---|---|---|
| Group I (very favorable) | solitary tumor <4DD | multiple tumors |
| II | 4-10DD | multiple 4-10DD |
| III | at equator or anterior | >10DD posterior |
| IV | multiple tumors, some >10DD | anteriorto ora serrata |
| V(least favorable) | massive tumor > ½ retina | vitreous seeding |
International Classification
| A | Small(<3mm) | >3mm from fovea | >1.5mm from optic nerve |
| B | (>3mm) | confined to retina | |
| C | Localizedvitreous or subretinal seeding. | <6mm from tumor | |
| D | Diffuse vitreous or subretinal seeding. | >6mm from primary | |
| E | No visual potential or tumor in anteriorsegment, in or onciliary body, neovascular glaucoma,vitreous hemorrhage, phthisical eye, orbital extension/proptosis. |
- Trilateral Retinoblastoma:
- bilateral intraocular retinoblastoma + ectopic intracranial retinoblastoma.
- Ectopic foci usually in pineal gland. (pinealoblastoma)
- Occurs in 2-5% with germline mutation.
- Usually pineal
- blastoma presents years after retinoblastoma, but may present before.
- Often show Flexner-Wintersteiner rosettes on histology.
- Embryological evidence for photoreceptor differentiation in pineal glando
- All retinoblastoma patients should undergo neuroimaging.
- Serial MRI with and without contrast = best (No radiation exposure)
- Decreased incidence of trilateral retinoblastoma with chemo –may be prophylactic.
- Median survival 8 month with CNS involvement.
- Prognosis:
- 95% survival if contained in eye
- Less than 50% survival if extraocular spread.
- Treatment:
- Goals
- #1 preserve life,
- #2 preserve eye,
- #3 preserve vision.
- Enucleation: definitive treatment. Usually enucleate if any of the following:
- Tumor >50% of eye
- Orbitalor ON involvement suspected
- Anterior segment involved
- Chemotherapy:
- carboplatin, vincristine, etoposide, cyclosporine
- IV treatment Q3-4 weeks in 4-9 cycles.
- Decrease tumor volume + finish treatment with laser, cryo, radiation.
- Periocular Chemo: in phase 1 + 2 trials, shows promise.
- Photocoagulation + hyperthermia:
- May kill tumor blood supply
- Cytotoxic effect of heat.
- Small tumors.
- Cryotherapy
- Small tumors <10mm diameter, <3mm thick.
- Anterior location (laser for posterior)
- Repetitive treatment+ close follow up.
- External Beam:
- retinoblastomais radiation responsive.
- 4,000 –4,500 cGy over 4-6 wks.
- Typically are bilateral not amenable to cryo or laser.
- 85% of globes retained.
- Visual function limited only by tumor location and secondary complications.
- Concerns:
- Retinoblastoma mutation associated with lifelong increased risk of osteosarcoma
- Risk of osteosarcoma is exacerbated by exposure to external beam radiation
- Midface hypoplasia
- Cataract
- Radiation optic
- Neuropathy + other radiation side effects.
- Combination therapy may decrease radiation and its secondary complications.
- Goals
- Brachytherapy: salvage therapy for some eyes
- Primary treatment for amenable eyes with small-medium tumors.
- Adenovirus targeted gene therapy in trials → makes tumor susceptible to ganciclovir –via Thymidine Kinase transfection
- Spontaneous Regression:
- May undergo complete necrosis + regression.
- Sometimes seen in phthisical eyes.
- Vitreous filled with islands of calcified cells in mass of fibrovascular connective tissue.
- Associated with exuberant proliferation of RPE + ciliary body
- Ghost contours of tumor cells.
- Prognosis:
- Survival greater than 95% with access to modern care.
- Extraocular extension is the most important poor prognostic factor
- Bilateral diseasemay have decreased survival secondary to optic intracranial foci
- Bilateral retinoblastoma leads to increased incidence of other tumors
- Mean time = 9 years
- 26% develop another tumor in 50 yrs.
- External beam radiation decreased latency and increased head + neck tumors.
- Most common secondary tumor = osteogenic sarcoma
- Also pinealoma, brain tumor, melanoma, sarcoma, primitive tumors
- 10-20% will develop tumor in 20 years
- 30-40% will develop 3rd tumor in 30 years
- Survival <50% in those who develop 2° osteogenic sarcoma.
Secondary Tumors
Metastaticintraocular tumors:
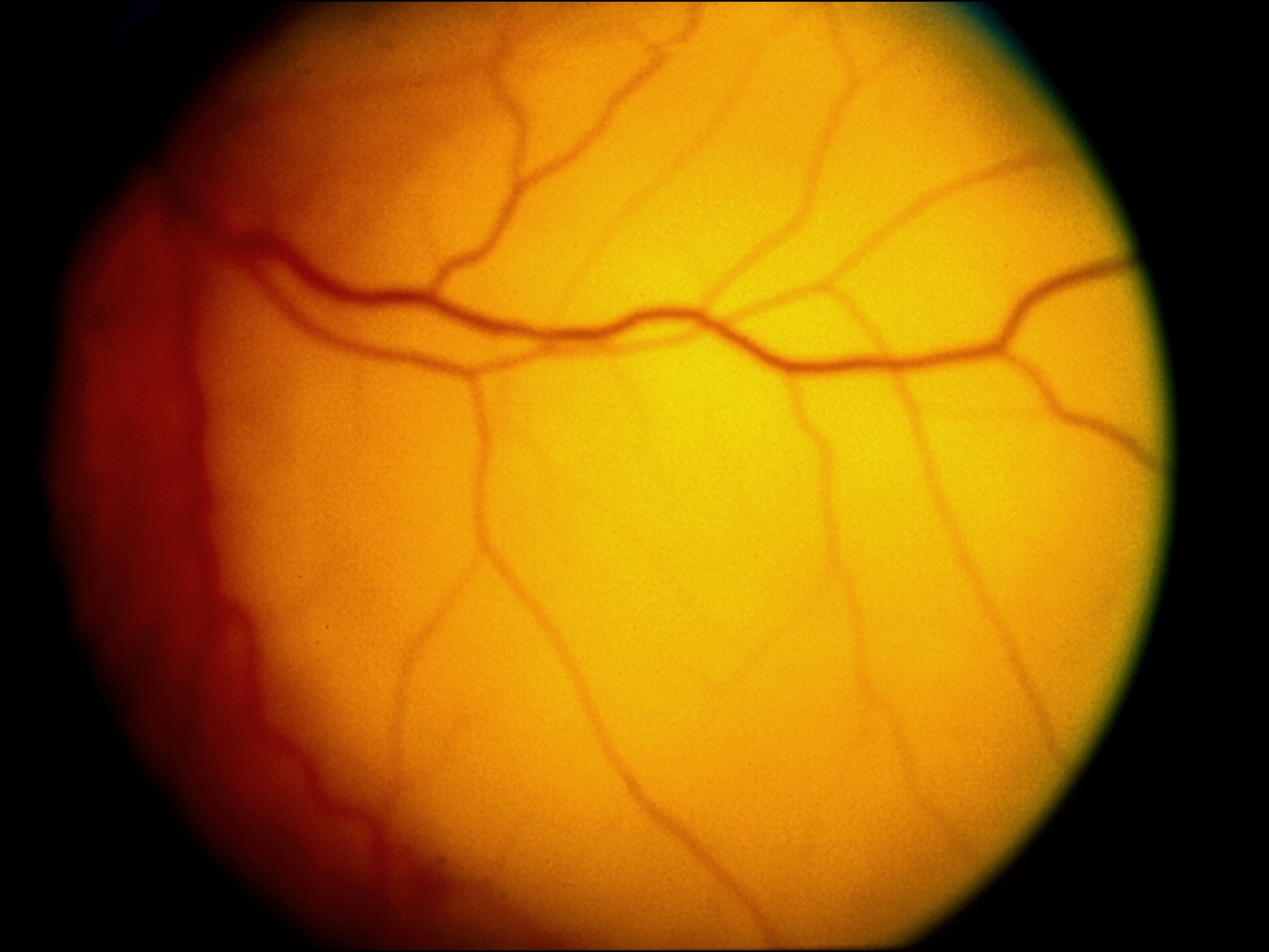
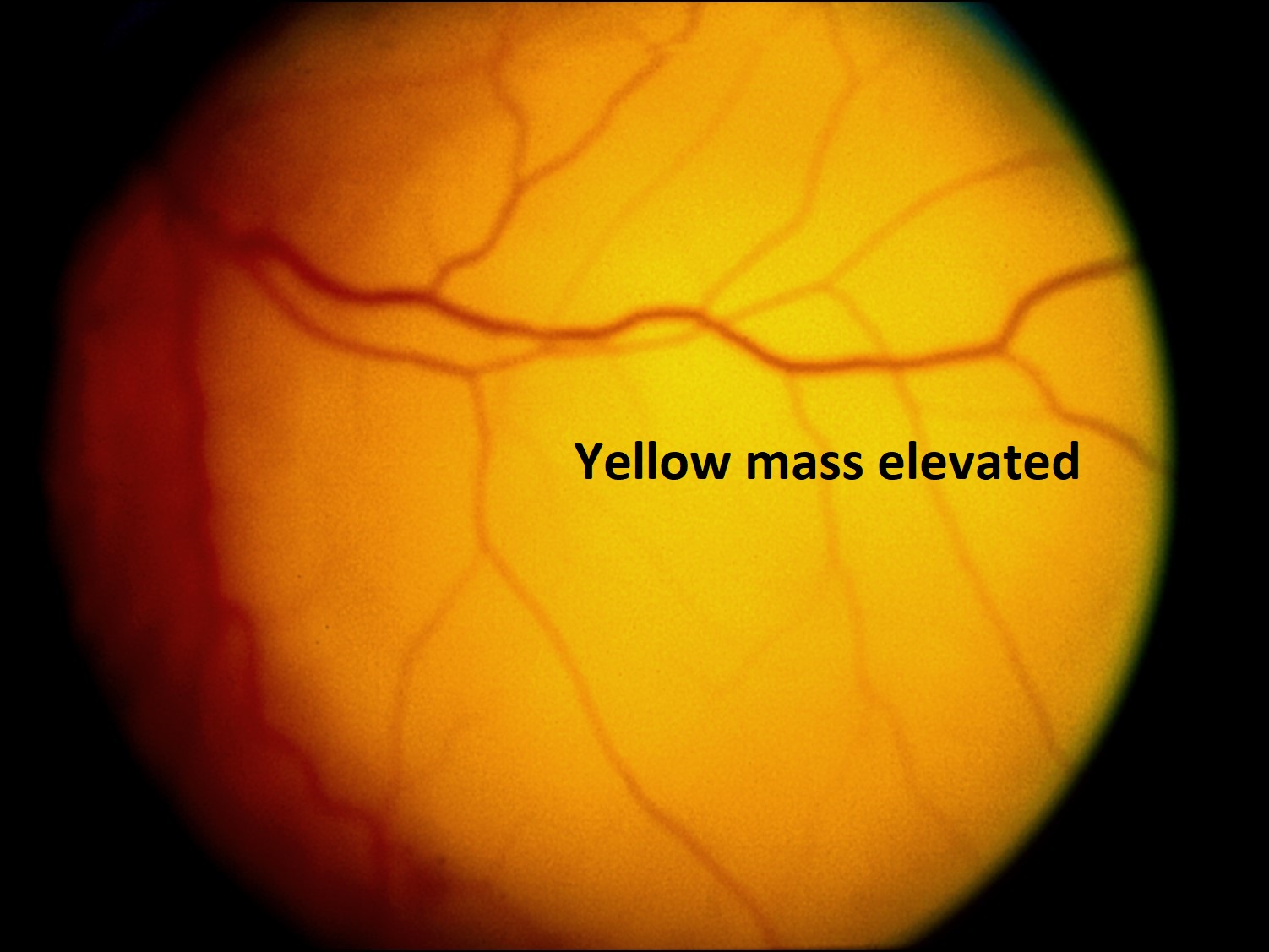
17.48 Fundoscopic photo
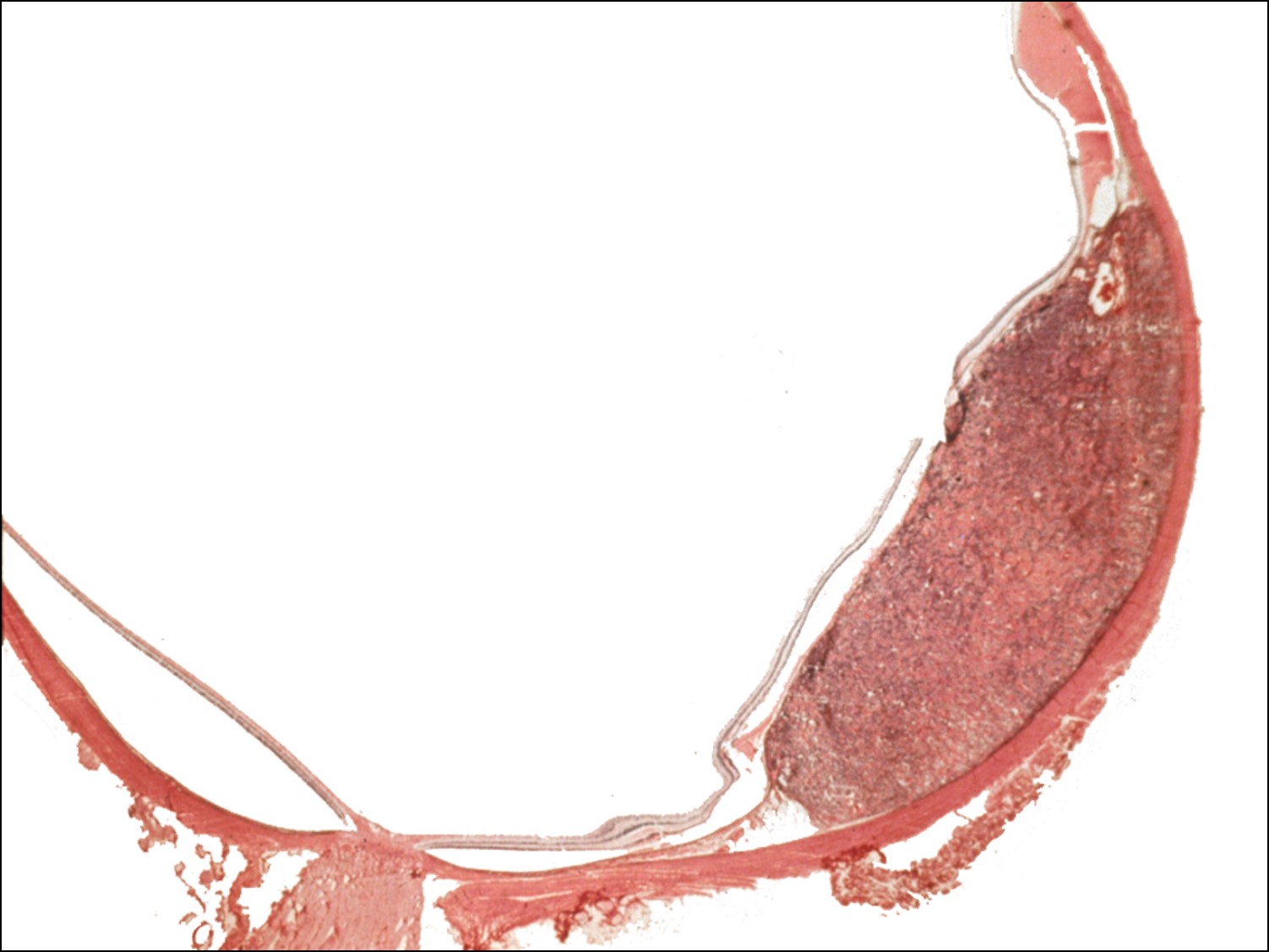
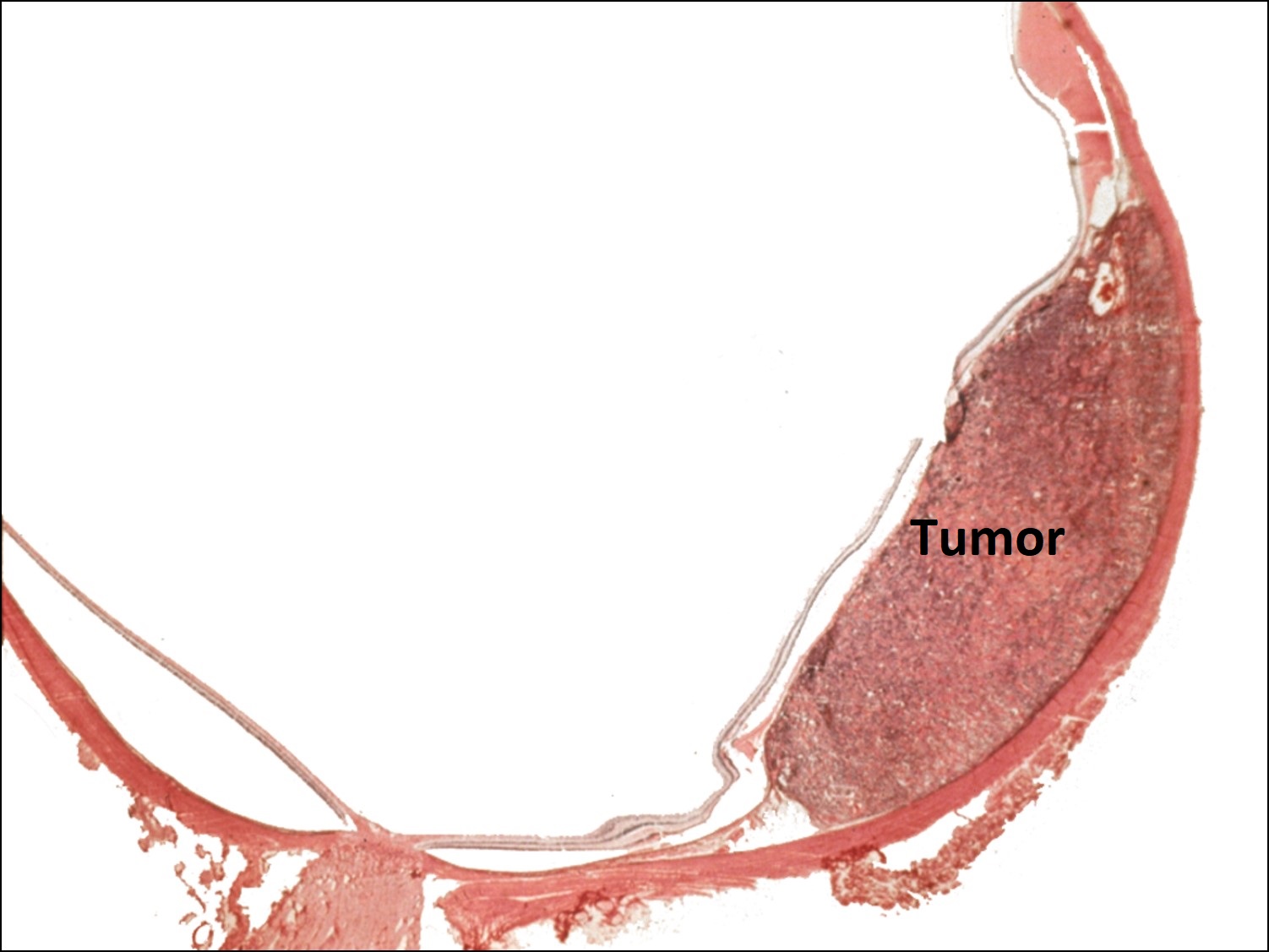
17.49 Lung cancer metastasized to choroid
Nick’s Tips:
- Metastases are the most common intraocular tumors in adults
- Metastases are the most common orbital tumors in adults
Mets to Eye
| Males | Females |
|---|---|
| Lung 40% | Breast 68% |
| Unknown 29% | Lung 12% |
| GI 9% | Unknown 12% |
| Kidney 6% | Others 4% |
| Prostate 6% | GI 2% |
| Skin 4% | Skin 1% |
| Others 4% | Kidney<1% |
- Choroid is 10-20 x more likely location for metastases, due to higher blood supply than iris or ciliary body
- Metastases to the retina + optic disc rare
- Bilateral in 20-25%
- Metastases are often yellow/white/grey, may have RPE changes
- Metastases to the posterior pole may cause decreased visual acuity
- Rarely break through Bruch’s membrane
- May cause retinal detachment over tumor
- Necrosis + uveitis possible
- Diagnosis:
- FA pattern of double circulation + early filling seen in choroidal melanomas is rare in metastatic tumors
- Ultrasound is helpful,
- A-scan shows moderate to high reflectivity (melanoma = low)
- Fine needle aspiration biopsy may help, but metastases may be too poorly differentiated to identify.
- Metastases to retina are very rare:
- Appear as white, non-cohesive lesions
- May appear similar to cotton wool spots
- May vitreous seed + appear like retinitis.
- Previous diagnosis versus unknown cancer diagnosis:
- Breast cancer
- 70-90% with metastatic ocular or orbital breast tumor to uvea have a history of breast CA diagnosis + treatment.
- If no prior diagnosis of breast cancer, then refer for breast exam
- Lung cancer
- Lung metastases often do not have prior diagnosis.
- Poor prognosis if uveal metastasis. Mean survival 1-67 mo
- Breast cancer
- Prognosis: Depends on primary tumor.
- Lung + GI is worst prognosis
- Carcinoma+ breast cancer is better prognosis
- Treatment: depends on many factors
- Goals
- Maintain vision
- Palliate pain.
- Types:
- Systemic chemo or hormone treatment
- Laser, cryo, beam radiation, etc….
- Enucleation with unrelenting pain.
- Goals
- Avenue of entrance to the orbit or globe
- Hematogenous inoculation is by far the most common
- Breast, lung, GI, and others
- Direct intraocular extension is rare because the sclera is a good barrier.
- Squamous cell carcinoma from conj. = most common
- Conj. Melanoma
- Basal cell of eyelid possible
- Hematogenous inoculation is by far the most common
Lymphomatous Tumors
Intraocular lymphoma:

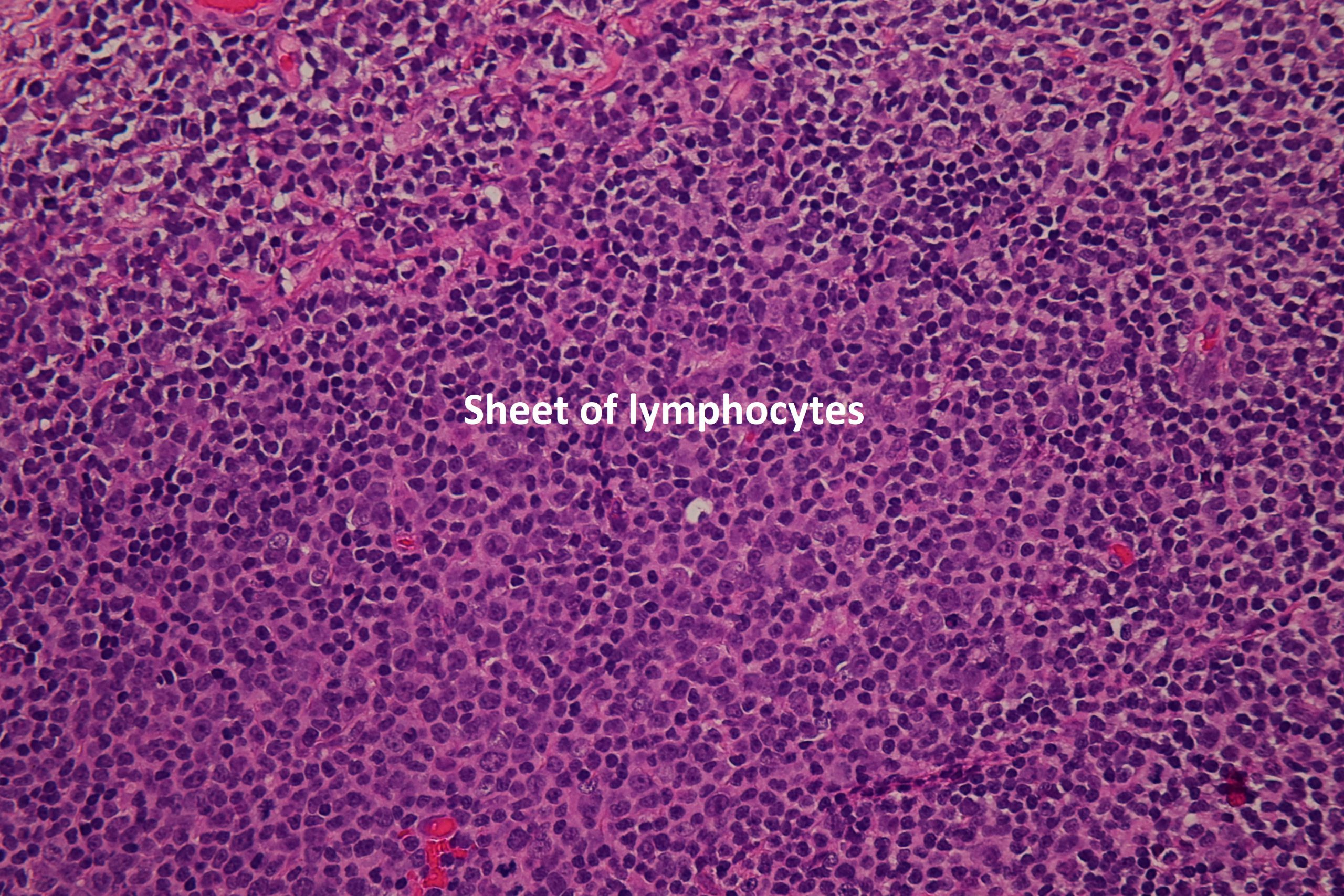
17.50 Lymphoma
- Type:
- Almost always Non-Hodgkin, B-cell, large cell.
- T-cell = rare.
- May invade any part of eye/orbit.
- Source:
- Most common = primary CNS lymphoma (25% have intraocular involvement) Vitreous + retina involved
- Rarely Systemic/visceral/nodal lymphoma: usually uveal tract + choroid.
- Making the diagnosis
- Any bilateral or chronic uveitis age over 50 should consider lymphoma.
- Diffuse vitreous cells, headlight in fog appearance.
- Complete neuro exam will be positive of deficiencies in 10% of patients
- 60% will have central nervous system involvement –should do
- CT
- MRI
- LP
- Diagnostic vitrectomy.
- Pathology
- Flow cytometry
- PCR
- Immunohistochemical stains
- Treatment
- Limited by blood-ocular barrier.
- Irradiation with external beam –tumor invariably reoccurs.
- Intraocular methotrexate → good response, low recurrence
- CNS treatment by oncologist
- Prognosis: poor, requires close f/u
Uveal Lymphoid Infiltration:
- Uveal Lymphoid Infiltration: (reactive lymphoid hyperplasia) –unknown etiology
- May occur at any site. –benign lymphoid infiltration
- Painless progressive vision loss
- Diffuse amelanotic thickening of choroid
- Exudative retinal detachment+ glaucoma in 85%, proptosis 15% due to orbit infiltration
- Treatment: high dose steroid, fractionated radiation
- Excellent prognosis
Leukemia
- Ocular involvement common –up to 80% on autopsy
- Up to 40% at diagnosis.
- Retinal lesions most commonly found, choroid is most common site.
- Choroid may act as sanctuary for leukemic cells
- Eye may be 1st sentinel of reactivation
- Ultrasound may be better than indirect ophthalmoscopy.
- May involve iris with small nodules @ pupil margin or diffuse thickens
- May have pseudohypopyonor 2° glaucoma.
- Retinal findings include:
- hard exudates
- CWS
- Pseudo-Roth spots.
- Yellow deposits in retina,
- Grey nodules in CML
- Perivascular streaks
- Vitreous involvement rare –use diagnostic vitrectomy if suspected
- Optic Nerve infiltration is anophthalmic emergency, treatment required immediately –treat with systemic + intrathecal chemo. +/-radiation
- Proptosis possible if orbital soft tissue infiltrates.
- May have opportunistic infections secondary to immunocompromise.
- Treatment = low dose radiation, systemic chemotherapy
Medulloepithelioma:
- Meulloepithelioma (diktyoma)
- Tumor of non-pigmented Ciliary Body epithelium
- Has benign + malignant forms
- Congenital
- Clinical diagnosis usually around age 4-12 years old. Sometimes diagnosed in adults
- Variably pigmented mass arising from ciliary body (rarely on orinretina)
- May erode into anterior chamber
- Treatment
- Enucleate–DO NOT SURGICALLY RESECT
- Observe
- Metastasisis RARE
- May treat small lesions with brachy therapy (I-125 plaque)
Leiomyomas, Neurilemomas, Neurofibromas:
VERY RARE: Usually misdiagnosed as amelanotic primary uveal melanomaand discovered on histopathology
References:
- Shields CL, Shields PW, Manalac J, Jumroendararasame C, Shields JA. Review of cystic and solid tumors of the iris. Oman J Ophthalmol, 2013 Sep-Dec; 6(3):159-164.
- Bianciotto C, Shields CL, Guzman JM, Romanelli-Gobbi M, Mazzuca D Jr, Green WR, Shields JA Assessment of anterior segment tumors with ultrasound biomicroscopy versus anterior segment optical coherence tomography in 200 cases. Ophthalmology. 2011 Jul; 118(7):1297-302.
- Shields CL, Arepalli S, Lally SE, Lally EB, Shields JA. Iris stromal cyst management with alcohol-induced sclerosis in 16 patients. JAMA Ophthalmol. 2014 Jun;132(6):703-8.
- Demirci H, Mashayekhi A, Shields CL, Eagle RC, Shields JA. Iris Melanocytoma: clinical features and natural course. Am J Ophthalmol. 2005 Mar; 139(3):468-75.
- Geisse LH, Robertson DM. Iris Melanoma. Am J Ophthalmol 1985. 99:638-648.
- Henderson E, Margo C. Iris Melanoma. Archives of Pathology & Laboratory Medicine. 2008 Feb 132(2):268-272.