
Orbital Rhabdomyosarcoma
Home / Basic Ophthalmology Review / Orbit
Topic: Orbital Rhabdomyosarcoma
Author: Colton McCoy, MBA, Texas Tech University Health Sciences Center School of Medicine
Overview:
Ocular RMS is a broad term that describes RMS occurring in the orbit, conjunctiva, anterior uveal tract, or eyelid. RMS can occur in many places throughout the body and comprises 4% of pediatric malignancies, with 10% of all RMS cases occurring within the orbit [3]. Around 250 new cases of RMS occur each year [3]. Rhabdomyosarcoma (RMS) is the most common primary malignant tumor of the orbit in children with a mean age of onset of 5-7 years old [5]. Although most cases of orbital RMS occur early in childhood, the disease has been reported from birth into the eighth decade of life. This is a life-threatening tumor and as such requires prompt diagnosis and treatment.
Orbital RMS can arise within the orbit (primary), can occur from direct extension from adjacent areas such as the nasopharynx (secondary), or after spread to the orbit from distant organs (metastatic). Primary orbital RMS was initially posited to originate from striated skeletal muscle of the orbit. However, it is now known that among the different histologic variants of RMS, the majority of cases find origin in pluripotent mesenchymal cells which retain the ability to differentiate into striated muscle [1]. This most common type of orbital RMS is referred to as embryonal RMS and is what will occupy the majority of this review.
Presentation:
Most cases of orbital RMS are located in the superonasal orbit. Thus, the hallmark presentation of orbital RMS is a rapidly occurring proptosis (80-100%) with inferotemporal displacement of the globe (70%) due to mass effect [4]. Patients may also present with blepharoptosis (30-50%), conjunctival and eyelid swelling (60%), palpable mass (25%), pain (10%), vision loss, or signs of sinusitis [3]. Rarely, orbital RMS can metastasize hematogenously to other organs (bone, bone marrow, lungs) or invade locally.

Figure 1: orbital mass, lateral view
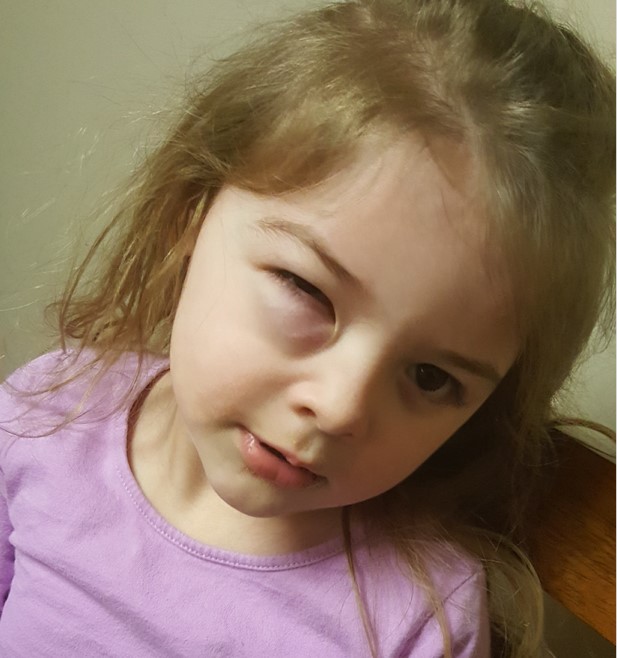
Figure 2: orbital mass, anterior view
Differential Diagnosis:
- Orbital Cellulitis
- Lymphangioma
- Idiopathic orbital inflammation
- Dermoid cyst
- Hemangioma
- Langerhans cell histiocytosis
- Other sarcomas
- Metastatic neuroblastoma
- Lymphoma
Diagnosis:
A thorough medical and ocular history, as well as imaging studies (especially CT or MRI) are prudent steps in the workup of suspected orbital RMS. CT imaging usually reveals a well-circumscribed, homogenous, round to ovoid mass which is isodense to muscle [3]. Similarly, on T-1 weighted MRI imaging, an orbital RMS will usually appear as a round or ovoid mass which is isointense compared to extraocular muscles and hypointense with respect to orbital fat. On T-2 weighted images the mass appears hyperintense to both extraocular muscles and orbital fat. Orbital RMS generally enhances markedly with gadolinium contrast. If orbital RMS is suspected on the basis of clinical and radiologic findings, prompt biopsy of the lesion should be performed. Histopathology, immunohistochemistry, and electron microscopy establish a definitive diagnosis of RMS. Tissue samples should be obtained by excisional biopsy or incisional biopsy with fine needle aspiration biopsy (FNAB) occasionally being performed.
Management:
There have been significant improvements in the early diagnosis and management of orbital RMS over the past 30 years. As recently as the early 1970s, the mortality rate associated with orbital RMS was 70%. Up until the 1960s, the treatment of choice for this condition was orbital exenteration. Nowadays, orbital RMS requires some combination of surgery, irradiation, and chemotherapy, depending on the intergroup rhabdomyosarcoma study group (ISRG) stage, with orbital exenteration being reserved for very severe cases. For these reasons, it is important that the pediatric oncologist becomes involved as early as possible. For embryonal RMS, the 5-year survival is as high as 94% with modern therapy [2]. Factors endowing patients with a more favorable prognosis include embryonal RMS, more favorable anatomic location, earlier stage, and patient age. Close follow-up is important to monitor for disease recurrence.
References:
- Karcioglu Z, Hadjistilianou D, Rozans M, et al. Orbital Rhabdomyosarcoma. Cancer Control2004;11:328-33.
- Kodet R, Newton WA Jr, Hamoudi AB, et al: Orbital rhabdomyosarcomas and related tumors in childhood: relation- ship of morphology to prognosis—an Intergroup Rhabdomyosarcoma study. Med Pediatric Oncol 29:51–60, 1997
- Shields J.A., Shields C.L. Rhabdomyosarcoma: review for the ophthalmologist. Surv Ophthalmol. 2003;48:39–57.
- Shields C, Shields J, Honavar S, et al. Clinical Spectrum of Primary Ophthalmic Rhabdomyosarcoma. Ophthalmology2001;108:2284-2292.
- Wharam M, Beltangady M, Hays D, et al. Localized orbital rhabdomyosarcoma. An interim report of the Intergroup Rhabdomyosarcoma Study Committee. Ophthalmology1987;94:251-4
Identifier: Moran_CORE_25481